Статья содержит краткий обзор клинических форм спинальной мышечной атрофии (СМА) и освещает результаты оригинального исследования по клинико-генетическим особенностям заболевания у детей в южных регионах Казахстана. Материалом исследования послужили 30 пациентов с симптомами СМА, отобранных в исследование в период с мая 2017 года по декабрь 2018 года. Проведено клинико-неврологическое и генетическое исследование на поиск мутаций в гене SMN1. Результаты исследования: 30 пациентов в возрасте от 1 месяца до 15 лет, проживающих в гг. Алматы, Шымкент и Алматинской, ЮжноКазахстанской/Туркестанской областях, мальчиков 13 (43,3%), Девочек 17 (56,7%).Клинико-генетический Диагноз «спинальная мышечная атрофия» впервые установлен у 16 детей (53,3%) и генетически подтвержден у 14 (46,7%). Ранжирование по типам заболевания: СМА 0 тип 1 (3,3%), СМА 1 тип 8 (26,7%), СМА II тип 14 (46,7%), СМА III тип 7 (23,3%). Большинство преДставлено пациентами коренной национальности 16 (53,3%). Семейный анамнез выявил инбреДный брак роДителей в 16,7% случаев и в 23,3% случаев отягощенность по СМА. Все пациенты на 1 гоДу жизни имели заДержку моторного развития различной степени тяжести: тяжелая степень заДержки отмечалась в 26,7%, среДняя в 46,6%, легкая в 26,7%. Навык самостоятельной хоДьбы изучался в группе Детей старше 1 гоДа: у 4 Детей (17,4%) развился в возрасте До 16 месяцев жизни, у 5(21,7%) развился в возрасте старше 16 месяцев, у 14 Детей (60,9%) навык самостоятельной хоДьбы не Достигнут. В неврологическом статусе преоблаДают симптомы поражения проксимальных групп мышц верхних и нижних конечностей в виДе мышечной слабости (96,7%, 100%), отсутствия коленных рефлексов (93,3%), снижение рефлексов с верхних конечностей (73,3%), фибрилляции языка (83,3%), патология брюшных рефлексов (100%). Генетический анализ метоДом ПЦР+ПДРФ выявил гомозиготную Делецию 7 и 8 экзонов гена SMN1 у 23 Детей (76,7%), гомозиготную Делецию 7 экзона 6 (20%), гомозиготную Делецию 8 экзона 1 (3,3%). ПозДняя Диагностика заболевания и отсутствие аДекватной реабилитационной помощи и мультиДисциплинарного ухоДа привели к утяжелению неврологического статуса и многочисленным осложнениям со стороны костно-суставной системы у пациентов исслеДуемой группы. Проблема СМА у Детей в Казахстане требуют Дальнейшего эпиДемиологического и клинического изучения на больших популяциях пациентов с анализом коррелятивных связей «фенотип-генотип» и применением современных Диагностических инструментов.
Введение. Спинальные мышечные атрофии (СМА) - термин, объединяющий наследственные нервно-мышечные заболевания, в патогенезе которых лежит первичное поражение клеток передних рогов спинного мозга и ядер мозгового ствола. СМАклассифицируют по типу наследования: аутосомно-рецессивные, аутосомно доминантные, Х-сцепленные и по преимущественной локализации мышечной слабости: проксимальные и дистальные формы. Аутосомно-рецессивные проксимальные СМА или 5q-SMA, встречаются до 95% всех случаев и обусловлены гомозиготной мутацией 7 и/или 8 экзонателомерной части гена SMN, расположенного в области хромосомы 5q12.2-q13.3. СМА относятся к орфанным заболеваниям с частотой заболевания 1 на 10 000 рожденных и с частотой гетерозиготного носительства в популяции 1 на 40-60 человек [1-3]. СМА являются наиболее распространенной генетической причиной младенческой смертности [4]. Проксимальные формыв зависимости от времени дебюта и симптомов заболевания подразделяются на 5 типов.
СМА I типа является наиболее тяжелым и распространенным клиническим вариантом, на который приходится около 50% всех пациентов с диагнозом «спинальные мышечные атрофии». Первые клинические признаки при этом типе появляются в возрасте до 6 месяцев, пациенты не приобретают способность сидеть без поддержки и обычно не выживают после первых 2 лет жизни. Неврологический статус характеризуются глубокой гипотонией, симметричным вялым параличом, отсутствием контроля головы, слабой спонтанной активностью. В наиболее тяжелых случаях слабые фетальные движенияпредполагают пренатальное начало заболевания с выраженной слабостью и контрактурами суставов при рождении. Эта форма была обозначена как СМА 0 тип. Клинически у всех детей с I типом заболеваниянаблюдается сочетание тяжелой гипотонии, арефлексии и симметричной мышечной слабости преимущественно в проксимальных отделах нижних конечностей, с сохранением силы и тонуса в лицевых мышцах. Поражение бульбарных мотонейронов вызывает фасцикуляции языка, слабоесосаниеи затрудненноеглотание, что со временемприводит к гастроэзофагальному рефлюксу, белково-энергетической недостаточности и аспирационной пневмонии.
СМА II типахарактеризуется дебютом в возрасте от 7 до 18 месяцев жизни. Пациенты достигают способности сидеть без поддержки, некоторые из них могут стоять, но не приобретают способность ходить самостоятельно. В неврологическом статусе: отсутствие сухожильных рефлексов, тремор верхних конечностей, контрактуры суставов и кифосколиоз. Спектр тяжести функциональных нарушений при II типе варьирует от слабых детей, которые могут только сидеть без поддержки и склонны к респираторным нарушениям и раннему сколиозу, до относительно более стабильных детей с более сильными мышцами туловища, конечностей и дыхательных путей. У слабых детей может развиться дыхательная недостаточность, требующая искусственной вентиляции легких.
СМА Штипа включает клинически гетерогенных пациентов, достигших все основные этапы моторного развития, в том числе, самостоятельной ходьбы. Однако, у большей части этих пациентов в раннем возрасте отмечалась проксимальная мышечная слабость. Степень функциональной недостаточности варьирует от тяжелых случаев, когда требуются вспомогательные средства передвижения, до легких, когда пациенты могут продолжать ходить и жить продуктивной жизнью с незначительной мышечной слабостью. У пациентов, которые теряют способность к передвижению, часто развивается сколиоз, ожирение и остеопороз, связанные с гиподинамией[2,3,5].
Традиционно при проведении ДНК-анализа в целях выявления мажорной гомозиготной делеции 7 и/или 8 экзона гена SMN1 применяется качественный простой и
Таблица 1 Характеристика двигательного и моторного развития пациентов с СМА
|
показатели |
характеристики |
n |
% |
|
Оценка двигательного развития на 1 году жизни(п=30) |
задержка моторного развития легкой степени |
8 |
26,7 |
|
задержка моторного развития средней степени |
14 |
46,6 |
|
|
задержка моторного развития тяжелой степени |
8 |
26,7 |
|
|
Развитие навыка самостоятельной ходь6ы(п=23) |
в возрастные сроки |
4 |
17,4 |
|
с задержкой |
5 |
21,7 |
|
|
не сформировался |
14 |
60,9 |
доступный метод полимеразной цепной реакции (ПЦР) и анализа полиморфизма длины рестрикционных фрагментов (ПЦР-ПДРФ-анализ). Для определения статуса гетерозиготного носителя и подсчета числа копий генов SMN1 и SMN2 необходимо использовать только количественный метод ДНК-диагностики, оптимальным из которых является мультиплексная лигазная реакция с последующей амплификацией (MLPA). В редких случаях возможно проведение поиска точковых мутаций методом прямого секвенирования гена SMN1 [6,7].
В Казахстане не проводились эпидемиологические и клинические исследования, посвященные спинальной мышечной атрофии у детей. Имеются отдельные публикации по клиническим случаям и генетической диагностике. Официальная статистическая отчетность по заболеваемости СМА не отражает действительное состояние вопроса. Актуальность исследования проксимальных спинальных мышечных атрофий у детей в южных регионах обусловлена значительным удельным весом наследственной нервно-мышечной патологии в структуре неврологической заболеваемости в этом регионе, большой частотой родственных браков и, возможно, высоким уровнем гетерозиготного носительства.
Цель: изучение клинико-генетических особенностей спинальной мышечной атрофии у детей в южных регионах Казахстана.
Материалы и методы исследования. Клиниконеврологическое и генетическое исследование 30 пациентов, находившихся на стационарном лечении в неврологических отделениях детских больниц гг.Алматы, Шымкент в период с мая 2017 года по декабрь 2018 года с клиническим диагнозом «спинальная мышечная атрофия, код M^-10G71.0». Отбор проводился из числа пациентов, направленных в стационар с симптомами неуточненной нервно-мышечной патологии, заддержки моторного развития и подозрением на спинальную мышеч ную атрофию, по критериям включения, утвержденным Международным консорциумом по СМА. Данные демографических показателей, семейного анамнеза, возраста и характеристики дебюта заболевания получены из медицинской документации и в процессе опроса.Проведено клинико-неврологическое обследование и ранжирование заболевания по типам в зависимости от возраста дебюта и клиники. Перед проведением параклинических исследований родители и доверенные лица несовершеннолетних пациентов дали письменное информированное согласие. Генетические исследования, направленные на поиск мутаций в гене SMN1 были проведены всем пациентам исследуемой группы в лаборатории МЦ «Центр молекулярной медицины», г.Алматы. Геномная ДНК для проведения анализа выделена из периферической крови стандартным методом. Для обнаружения мутаций применен метод ПЦР с последующим анализом ПДРФ с использованием ферментов рестрикции Bse 81 для 7 экзонаи Bse 1 для 8 экзона и регистрацией результатов в 7% полиакриламидном геле.
Результаты. Исследуемая группа представлена 30 пациентами в возрасте от 1 месяца до 15 лет включительно, среди них мальчиков - 13 (43,3%), девочек - 17 (56,7%), проживающих в гг. Алматы (10), Шымкент (4) и Алматинской (10), Южно-Казахстанской/Туркестанской областях (6). По этнической принадлежности группа разнородная: казахи - 16 (53,3%), узбеки - 5 (16,7%), уйгуры - 3 (10,0%), русские - 2 (6,7%), по 1 (2,5%) - представители других 4 этнических групп: татарин, дунганин, азербайджанец, украинец.
Изучение семейного анамнеза выявило наличие инбредного брака у родителей 5 (16,7%) пациентов, 25 (83,3%) опрошенных семей исключили наличие родственного брака. В 23,3% случаев (7 пациентов) выявлена отягощенность гередитарного анамнеза по СМА в виде наличия больных сибсов и больных родственников в семье. Опрос семей остальных 23 детей (76,7%) исключил наличие больных нервно-мышечной патологией в семье и среди родственников. Данных о гетерозиготном носительстве родителей пациентов нет.
Установлено, что пренатальный период был отягощен в 63,3% случаев в виде патологии беременности и задержки внутриутробного развития плода; на слабые фетальные движения указано в 11 случаях (36,7%); интранатальный период отягощен в 43,3% случаев в виде слабости родовой деятельности, применения родостимуляции и акушерских пособий. Неонатальный период у 9 детей (30%) характеризовался симптомами церебральной ишемии, синдромом угнетения. 3 детям (10%) исследуемой группы в раннем неонатальном периоде потребовалось применение ИВЛ. Слабое, неактивное сосание с первых дней отмечалось у 12(40%) детей, низкая прибавка в весе - у 14 (46,7%), ограничение двигательной активности - у 16 (53,3%), частыереспираторныезаболевания - у 12 (40%).
В таблице 1 дана характеристика раннего моторного развития детей исследуемой группы. Все пациенты на 1 году жизни имели задержку моторного развития различной степени тяжести: тяжелая степень задержки отмечалась у 8(26,7%) детей, средняя - 14(46,6%), легкая - 8(26,7%). Сроки формирования самостоятельной ходьбы изучался в группе детей старше 1 года, n=23. Навык самостоятельной ходьбы у 4 детей (17,4%) развился в возрастные сроки с 12 до 15 месяцев жизни, у 5 детей (21,7%) сроки формирования ходьбы установлены в возрасте старше 16 месяцев, у 14 детей (60,9%) навык ходьбы не был сформирован.
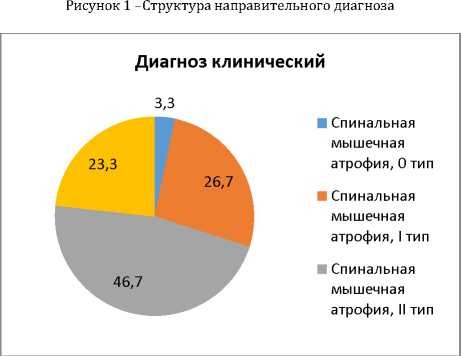
Особый клинический интерес представляет сопоставление направительного и клинического диагнозов. Структура направительного диагноза у пациентов исследуемой группы: «спинальная мышечная атрофия» - 14 детей (46,7%), «миопатия», «последствие натальной травмы шейного отдела позвоночника», «детский церебральный паралич, атонико-астатическая форма» - по 4 ребенка (13,3%), синдромом «вялого» ребенка - 2 детей (6,7%), по 1 ребенку (3,3%) с диагнозами«нейропатия постинфекционная», «задержка моторного развития центрального генеза» (Рисунок 1). Заболевание «СМА» у 14 детей выставлено клинически без генетического подтверждения и указано как синдром Верднига-Гоффмана. Структура заключительногоклинического диагноза состоит из 4 клинических типов СМА (Рисунок 2). В рамках исследования клинико-генетический диагноз «спинальная мышечная атрофия» впервые установлен у 16 детей (53,3%) и генетически подтвержден у 14 (46,7%).
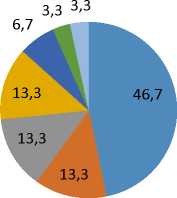
Диагноз направительный
- Спинальная мышечная атрофия
- Миопатия
- Натальная травма шейного отдела
- ДЦП
- Синдром вялого ребенка
- Нейропатия
Рисунок 2 - Структура заключительного диагноза
Все случаи СМА ранжированы по типам заболевания: СМА 0 тип - 1 (3,3%), СМА 1 тип - 8 (26,7%), СМА II тип - 14 (46,7%), СМА III тип - 7 (23,3%). Неврологический статус пациентов изучался по стандартной схеме неврологического осмотра с акцентом на нервно-мышечную и костно-суставную системы (Рисунок 3). В неврологическом статусе преобладают симптомы поражения проксимальных групп мышц верхних и нижних конечностей в виде мышечной слабости (96,7%, 100%), отсутствия коленных рефлексов (93,3%) и рефлексов с верхних конечностей (73,3%). Один из ранних локальных симптомов - фибрилляции языка
обнаружены в 83,3% случаев. У всех детей наблюдаются изменения со стороны брюшных рефлексов: снижение (56,7%) и отсутствие (43,3%). Данные результаты относятся к классическим симптомам проксимальной спинальной мышечной атрофии. Характерны изменения со стороны костно-суставной системы, как осложнения диффузной мышечной гипотонии, мышечной слабости, периферического пареза/паралича: кифосколиоз (84%), деформация грудной клетки (73,3%), патология стоп (93,3%), контрактуры суставов верхних (53,3%) и нижних конечностей (63,3%), деформация таза (38,5%).
Клинико-неврологический статус пациентов с СМА
патология стоп
контрактуры в суставах...
контрактуры в суставах. деформация таза
крыловидные лопатки деформация грудной клетки кифосколиоз
брюшные рефлексы -. СХР с верхних конечностей . СХР ахилловые - арефлексия СХР коленные - арефлексия
мышечная гипотония.
мышечная слабость.
фибрилляции языка
93,3%
63,3%
53,3%
38,5%
40,0%
73,3%
84,0%
43,3%
56,7%
73,3%
66,7%
93,3%
63,3%
90,0%
100,0%
96,7%
83,3%
Рисунок 3 Результаты клинико-неврологического осмотра детей с СМА
Таблица 2 Результаты генетической верификации(ПЦР+ПДРФ) пациентов с симптомами СМА
Все пациенты были обследованы комплексным методомПЦР+ПДРФ, направленным на поиск мутаций в гене SMN1. Результаты молекулярно-генетического
исследования представлены в таблице 2: гомозиготная
делеция 7 и 8 экзонов гена SMN1 обнаружена у 23 детей (76,7%), гомозиготная делеция 7экзона - 6 (20%),
гомозиготная делеция 8 экзона - 1 (3,3%).
Обсуждение и заключение. Полученные результаты показали некоторое преобладание девочек (17:13) в распределении по гендерному признаку, по этническому признаку - преимущественное преобладанием лиц казахской национальности (53,3%). Инбредный брак родителей выявлен в 16,7%, отягощенность семейного анамнеза по СМА - в 23,3%. У части пациентов в первых дней отмечались симптомы, которые можно рассматривать доклиническими и ранними клиническими признаками заболевания: слабое сосание (40%), ограничение активных движений (53,3%), низкая прибавка в весе (46,7%), частые респираторные заболевания (40%). Задержка в моторном развитии различной степени тяжести, обусловленная проксимальной слабостью, наблюдалась у всех 30 пациентов в раннем возрасте. Неврологический статус показал типичные для спинальной мышечной атрофии изменения в нервно-мышечной и костно-суставной с истемах. Высокий процент ошибочного д иагноза на амбулаторном уровне обусловлен низкой осведомленностью специалистов в вопросах клинической диагностики СМА и ограниченным доступом к генетическим исследованиям. Поздняя диагностика заболевания и отсутствие адекватной реабилитационной помощи и мультидисциплинарного ухода привели к утяжелению неврологического статуса и многочисленным осложнениям со стороны костносуставной с истемы у пациентов исследуемой группы. Спинальные мышечные атрофии у детей в Казахстане требуют дальнейшего эпидемиологического и клинического изучения на больших популяциях пациентов с анализом коррелятивных связей «фенотип-генотип» и применением современных диагностических инструментов.
|
n |
гомозиготная делеция 7 и 8 экзона |
гомозиготная делеция 7 экзона |
гомозиготная делеция 8 экзона |
|
|
СМА 0 |
1 |
1 (100,0%) |
||
|
СМА 1 |
8 |
8 (100,0%) |
||
|
ыСМА 2 |
14 |
11 (78,6%) |
2 (14,3%) |
1(7,1%) |
|
СМА 3 |
7 |
3 (42,9%) |
4 (57,1%) |
|
|
всего |
30 |
23 76,7% |
6 20,0% |
1 3,3% |
СПИСОК ЛИТЕРАТУРЫ
- Ingrid E. C. Verhaart, Agata Robertson, Ian J. Wilson, Annemieke Aartsma-Rus et al. Prevalence, incidence and carrier frequency of 5qlinked spinal muscular atrophy a literature review // Orphanet Journal of Rare Diseases. 2017. №12. Р. 124-126.
- Adele D'Amico, Eugenio Mercuri, Francesco D Tiziano, and Enrico Bertini. Spinal muscular atrophy // Orphanet J Rare Dis. 2011. №6. Р. 71-76.
- Mercuri et al. Diagnosis and management of spinal muscular atrophy: Part 1: Recommendations for diagnosis, rehabilitation, orthopedic and nutritional care // Neuromuscular Disorders. 2018. №28. Р. 103-115.
- Prior TW, Snyder PJ, Rink BD, Pearl DK, Pyatt RE, Mihal DC, Conlan T, Schmalz B, Montgomery L, Ziegler K, Noonan C, Hashimoto S, Garner S. Newborn and carrier screening for spinal muscular atrophy // Am J Med Genet A. 2010. №152. Р. 1605-1607.
- W David Arnold, DarineKassar, John T Kissel. Spinal muscular atrophy: diagnosis and management in a new therapeutic era // Muscle Nerve. 2015. №51(2). Р. 157-167.
- Arkblad EL, Darin N, Berg K, Kimber E, Brandberg G, Lindberg C, Holmberg E, Tulinius M, Nordling M. Multiplex ligation-dependent probe amplification improbe diagnostics in spinal muscular atrophy // Neuromuscul Disord. 2006. №16. Р. 830-838.
- Забненкова В.В., Дадали Е.Л., Поляков А.В. Проксимальная спинальная атрофия типов I-IV: особенности молекулярногенетической диагностики // Нервно-мышечные болезни. 2013. №3. С. 6-10.