В данном обзоре представлены молекулярно-генетические механизмы развития резистентности к инсулину в клетках, приводящей к сахарному диабету типа 2. Проанализирован сигнальный путь инсулина,и рассмотрено влияние мутаций на передачу сигналов и транспортировку глюкозы в клетку.
Введение.
Cахарный диабет -наиболее распространенное эндокринное заболевание человека, которое характеризуется комплексом нарушений в секреции и функциях инсулина, продуктивности глюкозы и т.д. Данное заболевание является основной причиной почечной недостаточности, слепоты, ампутации нижних конечностей и основным фактором развития сердечно-сосудистых заболеваний. Согласно национальному регистру РК больных сахарным диабетом, к концу 2017 года в Казахстане зарегистрировано около 309 тысяч людей с данным диагнозом, из которых около 2700 детей. С каждым годом число растет, к 2030 году количество лиц, страдающих диабетом, в стране может достигнуть 1 млн.
Основная причина появления сахарного диабета 2-го типа (СД2) - это инсулинорезистентность, то есть значительное снижение чувствительности клеток организма к воздействию инсулина. Нарушения происходят на ранних стадиях развития болезни. На молекулярном уровне многочисленные дефекты передачи сигналов инсулина влияют на резистентность к инсулину, уменьшают количество рецепторов инсулина, активность киназ рецепторов, фосфорилирование внутриклеточных субстратов, влияют на транслокацию и активацию транспортера глюкозы [1].
Большинство метаболических и антиапоптотических эффектов инсулина опосредуется сигнальным путем, который передается посредством мембранного рецептора. Сигнальный путь включает фосфорилирование белков субстрата рецептора инсулина (IRS), активацию фосфатидилинозитол-3-киназы (PI3-киназа), протеинкиназыВ (PKB или Akt) [2].
Рецептор инсулина.
Рецептор инсулина является тирозиновой протеинкиназой и состоит из двух a-субъединиц и двух ß-субъединиц, которые связаны дисульфидными связами [2].Синтез тетрамерной молекулы рецептора инсулина кодируется одной мРНК, и в результате трансляции образуется одна высокомолекулярная пептидная цепь.
Инсулин связывается с a-субъединицей рецептора и активирует тирозинкиназу в ß-субъединице. Когда тирозинкиназа активируется, она способствует аутофосфорилированию ß-субъединицы, так как для усиления активности киназы необходимо фосфорилирование трех главныхтирозиновых остатков (в позициях 1158, 1162, 1163) [2].
Мутации в гене рецептора инсулина были идентифицированы в нескольких редких формах тяжелой резистентности к инсулину, включая лепреханизм и синдром Рабсона-Менденхолла. Этим пациентам часто нужно в сотни раз больше инсулина, чем пациентам с обычной формой диабета [3, 4]. Большинство из этих пациентов имеют нонсенс или миссенс мутации во внеклеточном лиганд-связывающем домене или внутриклеточном тирозинкиназном домене рецептора, что приводит к значительно уменьшенному связыванию с инсулином, изменению кинетики связывания с инсулином или снижению активности тирозинкиназы, но некоторые также предполагают дефекты промотора, приводящие к уменьшению экспрессии мРНК рецептора [5, 6].
Также мутации в гене рецептора инсулина были идентифицированы в исследованиях, в которых изучали популяцию пациентов с сахарнымдиабетом типа 2. В этих исследованиях было показано варьирование мутаций в гене рецептора инсулина от 0,4% до 7,8%. Однако в большинстве исследований было исследовано только 20-30% белковой кодирующей последовательности гена рецептора инсулина. Кроме того, использованные методы были разработаны таким образом, чтобы идентифицировать определенные типы мутаций, и не могли идентифицировать делеции, мутации в промоторе или в других регуляторных областях гена рецептора инсулина[7].
Субстрат рецептора инсулина.
Тирозинкиназа рецептора инсулина фосфорилирует белки субстрата рецептора инсулина (IRS) по нескольким тирозиновым остаткам, что придает им способность соединяться с рядом белков, содержащих SH2-домены. К таким белкам в частности относится p85-субъединица PI3- киназы.Фосфорилирование IRS ведет к плейотропной реакции клетки на инсулиновый сигнал. От степени фосфорилирования субстрата зависит увеличение или уменьшение клеточного ответа на инсулин, амплитуда изменений в клетках и чувствительность к гормону. Лабораторные мыши, лишенные гена IRS1, проявляют резистентность к инсулину и сниженную толерантность при нагрузке глюкозой. Это указывает на то, что повреждения гена IRS1 могут быть причиной СД2[8].
Полиморфизм G971R вгене IRS1(rs1801278)наблюдается с более высокой частотой у пациентов с СД2 и приводит к снижению передачи сигналов инсулина, в основном снижая активность PI3-киназы [9, 10]. Хотя эти данные не были подтверждены в популяционныхисследованиях[11, 12], недавние исследования вновь подтвердили связь между полиморфизмом G971R в IRS1и СД2 [13, 14]. Также у пациентов с СД2 была обнаружена очень редкая миссенс мутация T608R(rs104893642)в IRS1, приводящяя к снижению передачи сигналов инсулина [15]. Многочисленные полиморфизмы были идентифицированы в гене IRS2 человека, но четкая связь между этими полиморфизмами и СД2 не обнаружена [16].
PI3-киназа.
Фермент PI3-киназа представляет собой гетеродимер, содержащий регуляторную (р85) и каталитическую (р110) субъединицы. В регуляторной субъединице есть два SH2- домена и SH3-домен, поэтому PI3-киназа с высоким сродством присоединяется к IRS1 [17]. Это приводит к активации каталитической субъединицы, которая быстро фосфорилируетфосфатидилинозитол 4,5-бисфосфат (PIP2) для получения фосфатидилинозитола 3,4,5-трифосфата (PIP3).
Различные изоформы регуляторной субъединицы PI3- киназы кодируются тремя различными генами. Pik3r1 кодирует 65-75% всех регуляторных субъединиц, в основном в форме p85α, а также формы p55α и p50α. Pik3r2 кодирует p85β и составляет ~ 20% регуляторных субъединиц. Pik3r3 кодирует p55γ, который по структуре похож на p55α, но выражен на низких уровнях в большинстве тканей.
Три различные каталитические субъединицы-p110a, ß и δ- производные трех разных генов. Связывание регулятора с каталитической субъединицей повышает стабильность каталитической субъединицы и поддерживает его в ингибированном состоянии. Это облегчается связыванием регуляторной субъединицы с конкретными мотивами фосфотирозина в белках IRS, что приводит к его активации [18-20]. Специфическое для печени отсутствие p110α и малое количество p110β у мышей приводит к непереносимости глюкозы и резистентности к инсулину [21, 22]. Удивительно, нокауты регуляторных субъединиц PI3- киназы, включая гетерозиготную делецию p85α, нокаутp85ßили p50a/p55a, демонстрируют повышенную чувствительность к инсулину [23, 24]. Недавно было показано, что p85α связывается с транскрипционным фактором XBP-1 и модифицирует развернутый белковый ответ, который способствует резистентности к инсулину [25, 26].
Полиморфизм M326I (rs3730089) в регуляторной субъединице p85aPI3-киназы был идентифицирован в ходе исследования популяции женщин племениПима и связан с уменьшением распространенности СД2[27]. Однако эта мутация M326I оказывает лишь незначительное влияние на сигнализацию инсулина in vitro, уменьшая связывание p85α с IRS1 и увеличивая деградацию p85α[28]. Другой полиморфизм в p85α SNP42(rs8192680) связан с гипергликемией натощак, но его молекулярный механизм пока неизвестен[29].
Мембрана PIP3 связывается с 3-фосфоинозитидзависимой протеинкиназой 1 (PDK1), которая содержит домен PH, и инициирует активацию PDK1. Известными субстратами PDK1 являются протеинкиназа B (PKB), а также атипичные формы протеинкиназы C (PKC) [30]. PDK1 фосфорилируетPKBнаThr-308 и активируетего [31]. Однако для полной активации требуется фосфорилирование PKB на Ser-473, это достигается вторым комплексом мишенейрапамицина у млекопитающих (mTORC2) [32, 33].
Протеинкиназа В.
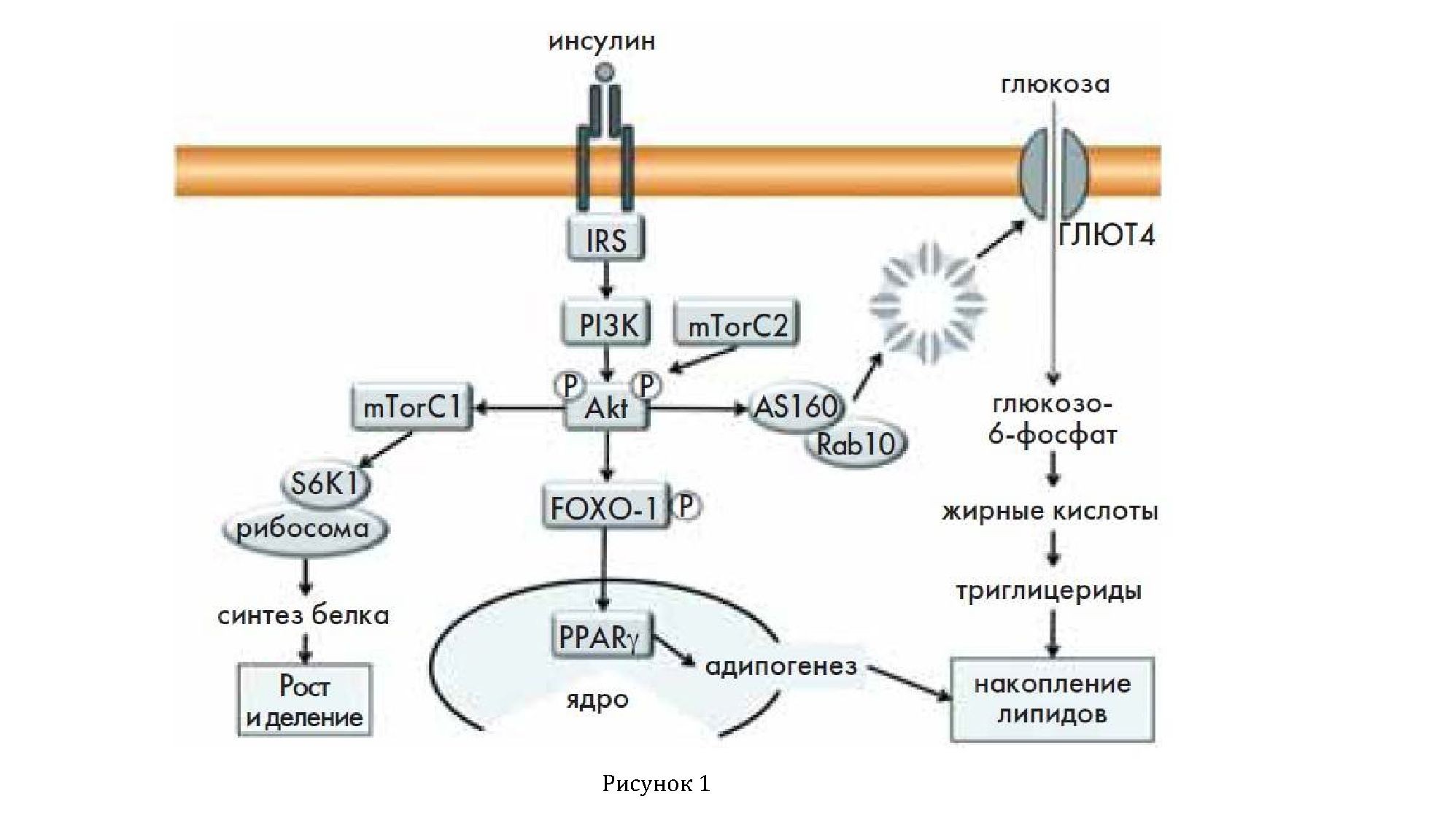
Активированная PKB фосфорилирует и активирует AS160 - фактор обмена гуаниловых нуклеотидов малых ГТФаз семейства Rab (рис. 1) [34]. Эти белки регулируют внутриклеточный транспорт везикул, а Rab10 отвечает за слияние GLUT4-содержащих везикул с мембраной клетки [35]. Таким путем PKB запускает экспонирование глюкозного транспортера и транспорт глюкозы в адипоциты и миоциты. В миоцитах глюкоза фосфорилируется и направляется на синтез гликогена, тогда как в адипоцитах она служит для липогенеза и из нее синтезируются триацилглицериды.
Инсулиновый сигнальный путь, показывающий связывание инсулина с IR, приводящий к активации GLUT4, который импортирует глюкозу в клетку. Связывание инсулина с IR активирует PI3-киназу, который продуцирует PIP2 и PIP3. Они служат связывающимисайтами для PDK1, который затем опосредуют активацию Akt. Увеличение количества свободных жирных кислот может индуцировать сериновое фосфорилирование белков IRS, что, в свою очередь, уменьшает фосфорилирование IRS-тирозина, тем самым снижая активность действия последующих эффекторных молекул данного пути.
IRS - белок рецептора инсулина; PI3K - фосфатидилинозитол-3-киназа; PDK1 - фосфоинотитозависимая киназа 1; Akt - протеинкиназа B; Foxo-1 - белок-фактор транскрипции; GLUT4 - транспортер глюкозы 4.
Класс белков PKB состоит из трех различных изоформ серин/треониновыхпротеинкиназ, кодируемых различными генами [36]. Все изоформы обладают доменом PH, позволяющим взаимодействовать с PIP3 и рекрутироваться в плазматическую мембрану.Данные, полученные на мышах с нокаутом гена PKB, дают четкий ответ на вопрос о том, требуется ли продукт гена PKB для нормального гомеостаза глюкозы. Хотя нарушение изоформы PKB1 у мышей не вызывало каких-либо значительных нарушений в метаболизме, мыши с отсутствием изоформы белка PKB2 показали резистентность к инсулину, в результате чего фенотип был очень похож на диабет типа 2 у людей [37, 38].
Впоследствии, последние исследования по наследственным мутациям пострецепторов инсулина у людей выявили миссенс-мутацию в гене киназного домена PKB2 у пациентов с тяжелой инсулинорезистентностью. Мутантная киназа не могла фосфорилировать нижестоящие мишени и опосредовать ингибирование фосфоенолпируваткарбоксикиназы (PEPCK), глюконеогенного ключевого фермента [39].
У пациентов с диабетом была обнаружена редкая миссенс мутация R274H(rs121434593) в PKB2, приводящая к потере активности киназы [40]. Две другие миссенс мутации R208K (rs35817154) и R467W (rs142926499) также были
идентифицированы у пациентов с диабетом, но, на удивление, in vitroэти мутантные формы проявляют неизмененную активность инсулин-индуцированной киназы[41]. У пациентов с диабетом типа 2 полиморфизм Q84R (rs2295490) в Trib3, приводящая к увеличению активности псевдокиназы,была связана с резистентностью к инсулину и снижением стимулированного инсулином фосфорилирования PKB[42, 43]. МутацияR363X (rs587777260)в AS160, приводящая к преждевременному стоп-кодону, была идентифицирована у пациента с тяжелой постпрандиальной гиперинсулинемией и действует доминантно-негативным образом для снижения транспорта глюкозы [44].
СПИСОК ЛИТЕРАТУРЫ
- Sesti G., Federici M., Lauro D., Sbraccia P., Lauro R. Molecular mechanism of insulin resistance in type 2 diabetes mellitus: role of the insulin receptor variant forms // Diabetes/Metabolism research and reviews. - 2001.- №17. - С. 363-373
- Saini V. Molecular mechanisms of insulin resistance in type 2diabetes mellitus // World J Diabetes. - 2010. - №1(3).- С. 68-75
- Kahn CR, Flier JS, Bar RS, Archer JA, Gorden P, Martin MM, Roth J 1976. The syndromes of insulin resistance and acanthosis nigricans. Insulin-receptor disorders in man // N Engl J Med. - 1976. - №294. - Р. 739-745
- Cochran E, Musso C, Gorden P 2005. The use of U-500 in patients with extreme insulin resistance // Diabetes Care. - 2005. - №28. - Р. 1240-1244.
- Taylor SI, Accili D, Cama A, Kadowaki H, Kadowaki T, Imano E, Sierra ML 1991. Mutations in the insulin receptor gene in patients with genetic syndromes of insulin resistance // Adv Exp Med Biol. -1991. - №293. - Р. 197-213
- Haruta T, Imamura T, Iwanishi M, Egawa K, Goji K, Kobayashi M 1995. Amplification and analysis of promoter region of insulin receptor gene in a patient with leprechaunism associated with severe insulin resistance // Metabolism. - 1995. - №44. - Р. 430-437.
- Taylor SI, Cama A, Accili D, et al. Mutations in the insulinreceptor gene // Endocrine Rev. - 1992. - №13. - Р. 566-595.
- Gerald M. Reaven, Ami Laws. Insulin Resistance: The Metabolic Syndrome X // Springer Science & Business Media. - 1999. - Р. 51-58.
- Almind K, Inoue G, Pedersen O, Kahn CR 1996. A common amino acid polymorphism in insulin receptor substrate-1 causes impaired insulin signaling. Evidence from transfection studies // J Clin Invest. - 1996. - №97. - Р. 2569-2575.
- Hribal ML, Tornei F, Pujol A, Menghini R, Barcaroli D, Lauro D, Amoruso R, Lauro R, Bosch F, Sesti G, et al. 2008. Transgenic mice overexpressing human G972R IRS-1 show impaired insulin action and insulin secretion // J Cell Mol Med. - 2008. - №12. - Р. 2096-2106.
- Florez JC, Sjogren M, Burtt N, Orho-Melander M, Schayer S, Sun M, Almgren P, Lindblad U, Tuomi T, Gaudet D, et al. 2004. Association testing in 9,000 people fails to confirm the association of the insulin receptor substrate-1 G972R polymorphism with type 2 diabetes // Diabetes. - 2004. - №53. - Р. 3313-3318.
- van Dam RM, Hoebee B, Seidell JC, Schaap MM, Blaak EE, Feskens EJ 2004. The insulin receptor substrate-1 Gly972Arg polymorphism is not associated with type 2 diabetes mellitus in two population-based studies // Diabet Med. - 2004. - №21. - Р. 752-758.
- Burguete-Garcia AI, Cruz-Lopez M, Madrid-Marina V, Lopez-Ridaura R, Hernandez-Avila M, Cortina B, Gomez RE, Velasco-Mondragon E 2010. Association of Gly972Arg polymorphism of IRS1 gene with type 2 diabetes mellitus in lean participants of a national health survey in Mexico: A candidate gene study // Metabolism. - 2010. - №59. - Р. 38-45.
- Martinez-Gomez LE, Cruz M, Martinez-Nava GA, Madrid-Marina V, Parra E, Garcia-Mena J, Espinoza-Rojo M, Estrada-Velasco BI, Piza- Roman LF, Aguilera P, et al. 2011. A replication study of the IRS1, CAPN10, TCF7L2, and PPARG gene polymorphisms associated with type 2 diabetes in two different populations of Mexico // Ann Hum Genet. - 2011. - №75. - Р. 612-620.
- Esposito DL, Li Y, Vanni C, Mammarella S, Veschi S, Della LF, Mariani-Costantini R, Battista P, Quon MJ, Cama A 2003. A novel T608R missense mutation in insulin receptor substrate-1 identified in a subject with type 2 diabetes impairs metabolic insulin signaling // J Clin Endocrinol Metab. - 2003. - №88. - Р. 1468-1475.
- Bernal D, Almind K, Yenush L, Ayoub M, Zhang Y, Rosshani L, Larsson C, Pedersen O, White MF 1998. IRS-2 amino acid polymorphisms are not associated with random type 2 diabetes amoung caucasians // Diabetes. - 1998. - №47. - Р. 976-979.
- Shaw LM 2011. The insulin receptor substrate (IRS) proteins: At the intersection of metabolism and cancer // Cell Cycle. - 2011. - №10. - Р. 1750-1756.
- Yu J, Zhang Y, McIlroy J, Rordorf-Nikolic T, Orr GA, Backer JM 1998. Regulation of the p85/p110 phosphatidylinositol 3′-kinase: Stabilization and inhibition of the p110α catalytic subunit by the p85 regulatory subunit // Mol Cell Biol. - 1998. - №18. - Р. 1379-1387.
- Burke JE, Vadas O, Berndt A, Finegan T, Perisic O, Williams RL 2011. Dynamics of the phosphoinositide 3-kinase p110δ interaction with p85α and membranes reveals aspects of regulation distinct from p110α // Structure. - 2011. - №19. - Р. 1127-1137.
- Zhang X, Vadas O, Perisic O, Anderson KE, Clark J, Hawkins PT, Stephens LR, Williams RL 2011. Structure of lipid kinase p110ß/p85ß elucidates an unusual SH2-domain-mediated inhibitory mechanism // Mol Cell. - 2011. - №41. - Р. 567-578.
- Jia S, Liu Z, Zhang S, Liu P, Zhang L, Lee SH, Zhang J, Signoretti S, Loda M, Roberts TM, et al. 2008. Essential roles of PI3K-p110β in cell growth, metabolism and tumorigenesis // Nature. - 2008. - №454. - Р. 776-779.
- Sopasakis VR, Liu P, Suzuki R, Kondo T, Winnay J, Tran TT, Asano T, Smyth G, Sajan MP, Farese RV, et al. 2010. Specific roles of the p110α isoform of phosphatidylinsositol 3-kinase in hepatic insulin signaling and metabolic regulation // Cell Metab. - 2010. - №11. - Р. 220230.
- Terauchi Y, Tsuji Y, Satoh S, Minoura H, Murakami K, Okuno A, Inukai K, Asano T, Kaburagi Y, Ueki K, et al. 1999. Increased insulin sensitivity and hypoglycaemia in mice lacking the p85 α subunit of phosphoinositide 3-kinase // Nat Genet. - 1999. - №21. - Р. 230-235.
- Ueki K, Yballe CM, Brachmann SM, Vicent D, Watt JM, Kahn CR, Cantley LC 2002. Increased insulin sensitivity in mice lacking p85β subunit of phosphoinositide 3-kinase // Proc Natl Acad Sci. - 2002. - №99. - Р. 419-424.
- Park SW, Zhou Y, Lee J, Lu A, Sun C, Chung J, Ueki K, Ozcan J 2010. The regulatory subunits of PI3K, p85α and p85β, interact with XBP-1 and increase its nuclear translocation // Nat Med. - 2010. - №16. - Р. 429-437.
- Winnay JN, Boucher J, Mori MA, Ueki K, Kahn CR 2010. A regulatory subunit of phosphoinositide 3-kinase increases the nuclear accumulation of X-box-binding protein-1 to modulate the unfolded protein response // Nat Med. - 2010. - №16. - Р. 438-445.
- Baier LJ, Wiedrich C, Hanson RL, Bogardus C 1998. Variant in the regulatory subunit of phosphatidylinositol 3-kinase (p85α): Preliminary evidence indicates a potential role of this variant in the acute insulin response and type 2 diabetes in Pima women // Diabetes. - 1998. - №47. - Р. 973-975.
- Almind K, Delahaye L, Hansen T, Van Obberghen E, Pedersen O, Kahn CR 2002. Characterization of the Met326Ile variant of phosphatidylinositol 3-kinase p85α // Proc Natl Acad Sci. - 2002. - №99. - Р. 2124-2128.
- Barroso I, Luan J, Middelberg RP, Harding AH, Franks PW, Jakes RW, Clayton D, Schafer AJ, O'Rahilly S, Wareham NJ 2003. Candidate gene association study in type 2 diabetes indicates a role for genes involved in β-cell function as well as insulin action // PLoS Biol. - 2002. - №1. - Р. 20-31.
- Kotani K, Ogawa W, Matsumoto M, Kitamura T, Sakaue H, Hino Y, Miyake K, Sano W, Akimoto K, Ohno S, Kasuga M. Requirement of atypical protein kinase clambda for insulin stimulation of glucose uptake but not for Akt activation in 3T3-L1 adipocytes // Mol Cell Biol. - 1998. - №18. - Р. 6971-6982.
- Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PR, Reese CB, Cohen P 1997. Characterization of a 3-phosphoinositide-dependent protein kinase, which phosphorylates and activates protein kinase Bα // Curr Biol. - 1997. - №7. - Р. 261-269.
- Sarbassov DD, Guertin DA, Ali SM, Sabatini DM 2005. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex // Science. - 2005. - №307. -Р. 1098-1101.
- Oh WJ, Jacinto E 2011. mTOR complex 2 signaling and functions // Cell Cycle. - 2011. - №10. - Р. 2305-2316.
- Watson RT, Pessin JE. Bridging the GAP between insulinsignaling and GLUT4 translocation // Trendsin Biochemical Sciences. -2006. - №31(4). - Р. 215-222.
- Leto D, Saltiel AR. Regulation of glucose transportby insulin: traffic control of GLUT4 // NatRev Mol Cell Biol. - 2012. - №13(6). - Р. 383396.
- Schultze SM, Jensen J, Hemmings BA, Tschopp O, Niessen M 2011. Promiscuous affairs of PKB/AKT isoforms in metabolism. //Arch Physiol Biochem. - 2011. - №117. - Р. 70-77.
- Cho H, Thorvaldsen JL, Chu Q, Feng F, Birnbaum MJ. Akt1/PKBalpha is required for normal growth but dispensable formaintenance of glucose homeostasis in mice // J Biol Chem. - 2001. - №276. - Р. 38349-38352.
- Cho H, Mu J, Kim JK, Thorvaldsen JL, Chu Q, Crenshaw EB 3rd, Kaestner KH, Bartolomei MS, Shulman GI, Birnbaum MJ. Insulin resistance and a diabetes mellitus-like syndromein mice lacking the protein kinase Akt2 (PKB beta) // Science. - 2001. - №292. - Р. 1728-1731.
- George S, Rochford JJ, Wolfrum C, Gray SL, Schinner S,Wilson JC, Soos MA, Murgatroyd PR, Williams RM, AceriniCL, Dunger DB, Barford D, Umpleby AM, Wareham NJ,Davies HA, Schafer AJ, Stoffel M, O'Rahilly S, Barroso I. Afamily with severe insulin resistance and diabetes due to amutation in AKT2 // Science. - 2004. - №304. - Р. 1325-1328.
- George S, Rochford JJ, Wolfrum C, Gray SL, Schinner S, Wilson JC, Soos MA, Murgatroyd PR, Williams RM, Acerini CL, et al. 2004. A family with severe insulin resistance and diabetes due to a mutation in AKT2 // Science. - 2004. - №304. - Р. 1325-1328.
- Tan K, Kimber WA, Luan J, Soos MA, Semple RK, Wareham NJ, O'Rahilly S, Barroso I 2007. Analysis of genetic variation in Akt2/PKB-ß in severe insulin resistance, lipodystrophy, type 2 diabetes, and related metabolic phenotypes // Diabetes. - 2007. - №56. - Р. 714-719.
- Prudente S, Hribal ML, Flex E, Turchi F, Morini E, De Cosmo S, Bacci S, Tassi V, Cardellini M, Lauro R, et al. 2005. The functional Q84R polymorphism of mammalian Tribbles homolog TRB3 is associated with insulin resistance and related cardiovascular risk in Caucasians from Italy // Diabetes. - 2005. - №54. - Р. 2807-2811.
- Prudente S, Scarpelli D, Chandalia M, Zhang YY, Morini E, Del Guerra S, Perticone F, Li R, Powers C, Andreozzi F, et al. 2009. The TRIB3 Q84R polymorphism and risk of early-onset type 2 diabetes // J Clin Endocrinol Metab. - 2009. - №94. - Р. 190-196.
- Dash S, Sano H, Rochford JJ, Semple RK, Yeo G, Hyden CS, Soos MA, Clark J, Rodin A, Langenberg C, et al. 2009. A truncation mutation in TBC1D4 in a family with acanthosis nigricans and postprandial hyperinsulinemia // Proc Natl Acad Sci. - 2009. - №106. - Р. 9350-9355.