Данная статья посвящена одному из направлений в детской неврологии – нервномышечным заболеваниям или в англоязычной литературе все чаще можно встретить термин «миология». Заболевания, описываемые в статье, относятся к категории орфанных, в лечении которых используются высокотехнологичные и дорогостоящие методы генной инженерии. В статье представлены основные трудности и вопросы, требующие анализа и решения, а также предложены возможные варианты управления этой проблемой.
Ключевые слова: нервно-мышечные заболевания, спинальная мышечная атрофия, миодистрофия Дюшена, мультидисциплинарный подход к диагностике, лечение, менеджмент.
Нервно-мышечные заболевания это большая группа заболеваний, в основе которых лежит нарушения морфологии или функции нервно-мышечного аппарата. В основе патогенеза этих заболеваний лежит генетический дефект, отвечающий за формирование и функционирование передних рогов спинного мозга, мышц, периферических нервов или синапса. Согласно анатомическому субстрату определяется четыре большие группы нервно-мышечных заболеваний.
Так при повреждении передних рогов спинного мозга возникает такое заболевание как спинальная мышечная атрофия в основе патогенеза, которого лежит аутосомно-рецессивная мутация в одном генетическом локусе на коротком плече 5-й хромосомы, приводящая к гомозиготной делеции в гене SMN1 и SMN2.
Спинальная амиотрофия (СМА) I типа начинается еще до рождения и проявляется в возрасте до 6 мес. Отмечается мышечная гипотония (часто заметная при рождении), гипорефлексия, фасцикуляции языка и выраженные затруднения при сосании, глотании и дыхании. Смерть наступает от дыхательной недостаточности на первом году жизни в 95% случаев, и к 4 годам погибают все больные. При СМА типа II симптомы обычно проявляются в возрасте 3–15 мес.; < 25% заболевших детей способны сидеть, но они не могут ходить или ползать. Развивается вялый паралич и фасцикуляции, что трудно выявить у маленьких детей. Выпадают глубокие сухожильные рефлексы. Может наблюдаться расстройство глотания. Большинство детей к 2-3 годам становятся прикованными к инвалидной коляске. Заболевание часто приводит к смерти в раннем возрасте от дыхательных осложнений.
Однако прогрессирование заболевания может внезапно остановиться, но устойчивая слабость и высокий риск тяжелого сколиоза и его осложнений сохраняются. СМА III типа обычно проявляется в возрасте между 15 мес. и 19 годами. Признаки похожи на симптомы I типа заболевания, но болезнь прогрессирует медленнее, а продолжительность жизни больше (иногда нормальна). Некоторые семейные случаи связаны с ферментными дефектами (например, дефицит гексозаминидазы).
Симметричная слабость и атрофии, начинаясь с четырехглавой мышцы бедра и сгибателей бедра, постепенно распространяются дистальнее, становясь наиболее выраженными на голенях. Позже поражаются руки. Продолжительность жизни зависит от развития дыхательных осложнений. СМА IV типа может наследоваться по рецессивному, доминантному или Х-сцепленному типу; с дебютом в зрелом возрасте (30–60 лет) и медленно прогрессирующей слабостью и атрофией в основном проксимальных мышц. Это заболевание трудно отличить от амиотрофического бокового склероза, поражающего главным образом нижние мотонейроны [1,2,3].
При первичном повреждении мышц выделяют следующие виды, такие как мышечные дистрофии, которые характеризуются специфическими аномалиями мышечного волокна, которые обнаруживаются при мышечной биопсии (например: изменением размеров мышечного волокна, появлением некротических изменений в мышечном волокне, формирование рубцовых 12
ОҢТҮСТІК ҚАЗАҚСТАН МЕДИЦИНА АКАДЕМИЯСЫ, ХАБАРШЫ №1(95), 2022 изменений в мышечном волокне или воспалительных реакций). Примерно 30 различных генетических нарушений лежит в основе патогенеза мышечных дистрофинопатий. Дистрофинопатии это спектр мышечных заболеваний, причиной которых являются различные повреждения в гене дистрофина.
Самым тяжелым заболеванием из этого спектра является миодистрофия Дюшена, причиной которого является отсутствие полноценного белка дистрофина. Снижение или укорочение белка дистрофина ассоциируется с менее тяжелой формой болезни и называется миодистрофия Беккера. Клиническим признаками миодистрофии Дюшена является слабость и исчезновение различных групп мышц.
В более поздних этапах заболевания вовлекаются мышцы сердца и желудочно-кишечного тракта. Причиной является мутация в гене DMD на Х хромосоме. Этот ген регулирует продукцию белка, который называется дистрофин и входит в состав внутреннего слоя мембраны скелетных и сердечной мышц. Дистрофин играет важную роль в поддержании мембраны (сарколеммы) в мышечных клетках.
Мышечная дистрофия Дюшена обычно проявляется в раннем возрасте 2-4 года, то есть тогда, когда ребенок начинает ходить. Больные мальчики развивают мышечную слабость и атрофию проксимальных мышц, таких как бедра и мышцы таза, плечи и мышцы плечевого пояса. Однако, другие группы мышц имеют псевдогипертрофию. По мере прогрессирования заболевания мышечная слабость и атрофия распространяется на дистальные группы мышц, икроножные, предплечья, шею и туловище [4,5].
Также при повреждении синапса наблюдается другая группа заболеваний, называющихся миастении. Миастения гравис это астенический бульбарный паралич, характеризуется выраженной слабостью и утомляемостью мышц. При этом заболевании поражаются холинорецепторы постсинапитической мембраны. В процесс может вовлекаться любая мышца тела, однако имеется тенденция к преимущественному поражению мышц лица, тела, губ, глаз, языка, глотки и шеи. Причины заболевания не всегда удается установить, так как она может иметь наследственный характер или аутоиммунный. Нередко миастения сочетается с гиперплазией или опухолью вилочковой железы. Иногда миастенические синдромы могут сопровождать органические заболевания нервной системы такие как боковой амиотрофический склероз, поли и дерматомиозиты, а также рак легкого, молочной железы, яичника, представительной железы. Женщины болеют чаще, болезнь начинается в возрасте 20-30 лет, но может и раньше. Аутоиммунный характер этого заболевания доказывает обнаружение аутоантител, антител к рецепторам постсинаптической мембраны, приводящих к деструкции и блоке передачи нервного импульса[7].
При повреждениях нервов наблюдается группа заболеваний, называемых невропатиями, которых очень много в зависимости от группы периферических нервов, которые подвержены патологическому процессу. В детской практике наиболее часто встречаются наследственные сенсорные и моторные полинейропатии. Врожденные невропатии и невропатии раннего возраста представляют собой группу редких и сложных состояний с широким фенотипическим и генетическим разнообразием. Чаще всего клинически они представлены такими заболеваниями как наследственные моторные сенсорные невропатии (НМСН) или болезнь Шарко-Мари-Тус (ШМТ), наследственные моторные невропатии (НМН) и наследственные сенсорные невропатии (НСН). Нейрофизиологическая классификация: демиелинизирующие – тип I и аксональные – тип II. [6]
Таким образом, в данном обзоре будет представлена проблема диагностики, лечения и ведения пациентов с нервно-мышечными заболеваниями, которые сами по себе являются редкими, то есть встречаются 1 на 10.000 населения, но в совокупности могут составлять доставочной большей процент болеющего детского населения. Актуальность поднимаемой проблемы еще обусловлена тем, что эти заболевания приводят к инвалидизации и очень сильно могут сокращать продолжительность жизни детей и без своевременного и корректного лечебно-диагностического и реабилитационного алгоритмов медицинское и социальное сопровождения таких пациентов очень проблематично.
В данной публикации хотелось бы больше обсудить вопросы наиболее часто встречающихся форм нервно-мышечных заболеваний, таких как Спинальная мышечная атрофия и Прогрессирующая мышечная дистрофия Дюшена/Беккера.
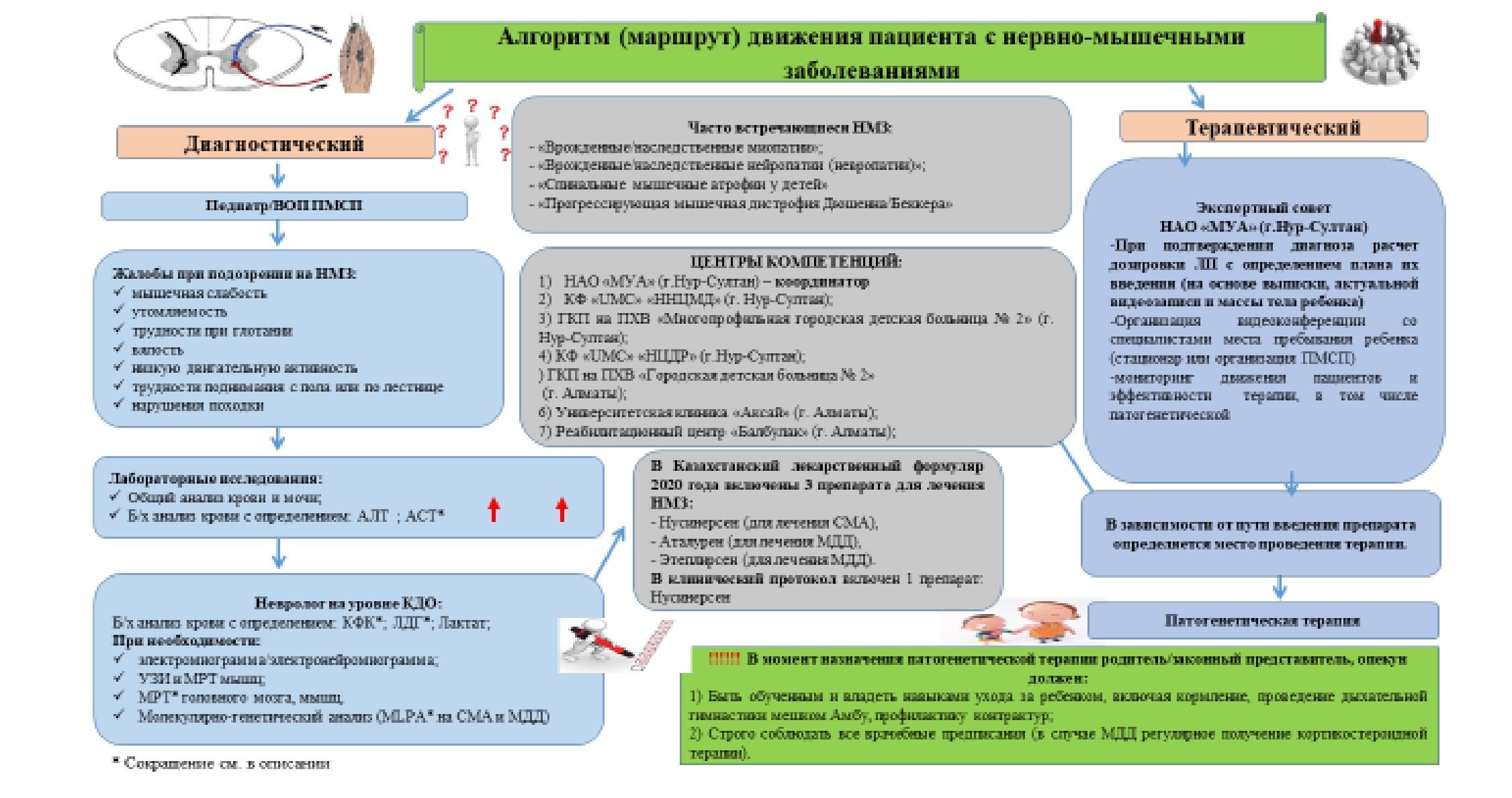
Данные заболевания дебютируют в раннем детском возрасте, приводят к быстрой инвалидизации и влияют на продолжительность жизни детей. В последние пять –шесть лет эти заболевания переведены из ранга неизлечимых в категорию курабельных состояний, поэтому своевременность и корректность проведения лечебно-диагностических процедур может сильно повлиять на прогноз этих заболеваний, а также повлиять на статистические показатели младенческой и детской инвалидности и смертности.
Также к возникающим вопросам на сегодня можно отнести не только отсутствие полноценного регистра пациентов, но и низкую информированность врачей неврологов , а также смежных специальностей об особенностях данной нозологии, недостаточность развития инфраструктуры организации помощи пациентам (отсутствие готовности медицинских учреждений к оказанию помощи пациентам данного профиля, в виду отсутствия утвержденных нормативно-правовых актов, регламентирующих работу с данным профилем пациентов). Кроме того, важным моментом является низкая информированность населения о данном заболевании и высокая стоимость лечения. Так самым дорогим медицинским препаратом в мире на сегодня является препарат генной терапии – онасемноген абепарвовек (золгенсма), который используется для лечения детей со спинальной мышечной атрофией до 2 лет.
Таким образом, в 2020 году был утверждён алгоритм движения пациентов с нервномышечными заболеваниями, который помогает оптимизировать процесс. В детской практике. Однако в ходе работы с данным контингентом появилась необходимость обсуждения вопроса организации помощи взрослым пациентам. Поскольку за последние два года данный алгоритм показал свою жизнестойкость, поэтому данная модель рекомендуется к рассмотрению и специалистами взрослыми неврологами.
Безусловно, в обсуждении вопроса об адекватной, своевременной лечебнодиагностической помощи, мы должны обсудить вопрос генетической диагностики, так как она лежит в основе не только подтверждения диагноза, но и выбора патогенетического лечения, который напрямую зависит от типа мутации, лежащей в основе заболевания.
Таким образом, исходя из сложившейся ситуации в Казахстане мы можем обсуждать следующие варианты генетической диагностики как количественная диагностика, методом MLPA: определение делеций 7 и/или 8 экзона в гене SMN 1 и подсчет числа копий гена SMN1, SMN2 (проводиться в генетической лаборатории КФ UMC Национальный центр материнства и детства, г.Нур-Султан) в рамках ГОБМП, бесплатно для пациента, а также в генетической лаборатории
14больницы УДП г. Нурсултан. Также с 2021 года в стране работает горячая линия- 7 775 886 87 23 по логистическому сопровождению биоматериала пациентов в генетические лаборатории генетической диагностики, поддержанная ОО «Общество детских неврологов, нейрофизиологов, психиатров и психотерапевтов» (https://neurosociety.kz/) так как это оптимизирует диагностический процесс и демонстрирует максимально дружелюбное отношение к нуждам маленького пациента и его семьи, позволяя избежать не всегда удобное путешествие в г. НурСултан или г. Алматы из дальних регионов Казахстана.
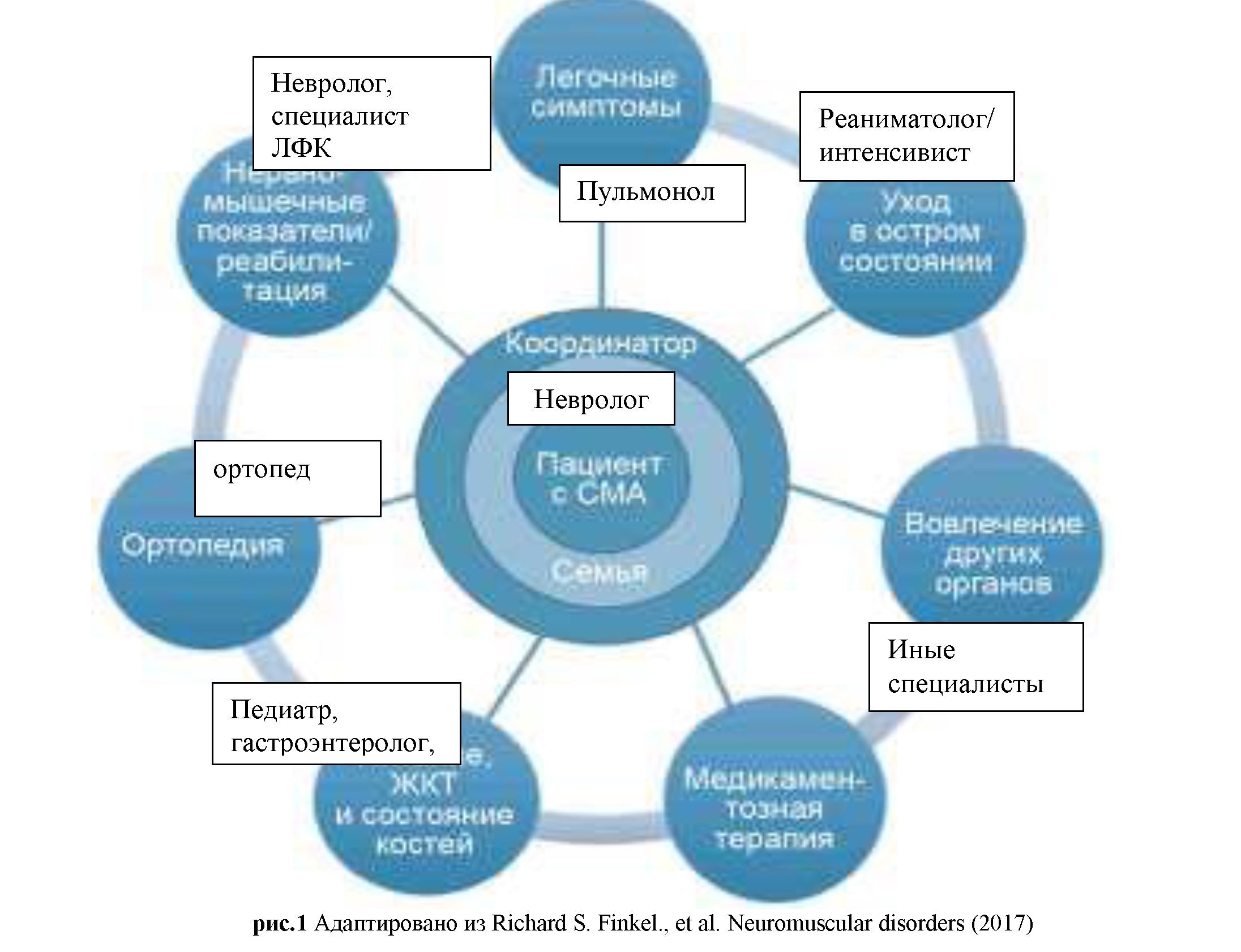
Наконец, обсуждая вопрос организации помощи детскому населению необходимо стремиться к организации мульти дисциплинарной помощи по модели успешно применяемой в других странах.
Анализируя представленную выше информацию о присутствии проблемы диагностики нервно-мышечной патологии, а также констатируя факт высокой специфичности данных заболеваний, сложность и неоднозначность интерпретации генетических диагностических данных, а также выработанные на сегодня патогенетические генно-инженерные методики коррекции заболеваний. Это дает основание вывести их в разряд орфанных, высокоспециализированных, требующих четких отлаженных действий мультидисциплинарной команды по оказанию помощи, а также финансово-затратных нозологий. Решение вопроса поддержания и развития таких пациентов должно приниматься экспертной группой, в основе решения которой должны лежать не только особенности генетической природы заболеваний и потенциальные возможности коррекции, но также высокая комплаентность и приверженность пациентов и их родителей к лечению и жизни с такими особенными состояниями.
 15
15
- Mercuri et al. Diagnosis and management of spinal muscular atrophy: Part 1: Recommendations for diagnosis, rehabilitation, orthopedic and nutritional care// Neuromuscular Disorders 28 (2018) 103–115
- Finkel et al. Diagnosis and management of spinal muscular atrophy: Part 2: Pulmonary and acute care; medications, supplements and immunizations; other organ systems; and ethics// Neuromuscular Disorders 28 (2018) 197–207
- Birnkrant DJ, Bushby K, Bann CM, Alman BA, Apkon SD, Blackwell A, Case LE, Cripe L, Hadjiyannakis S, Olson AK, Sheehan DW, Bolen J, Weber DR, Ward LM; DMD Care Considerations Working Group. Diagnosis and management of Duchenne muscular dystrophy, part1: diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol 2018;17:251-267. doi: 10.1016/S1474-4422(18)30024-3. Epub 2018 Feb 3.
- Birnkrant DJ, Bushby K, Bann CM, Alman BA, Apkon SD, Blackwell A, Case LE, Cripe L, Hadjiyannakis S, Olson AK, Sheehan DW, Bolen J, Weber DR, Ward LM; DMD Care Considerations Working Group Diagnosis and management of Duchenne muscular dystrophy, part 2: respiratory, cardiac, bone health, and orthopaedic management. Lancet Neurol. 2018 Apr;17(4):347-361. doi: 10.1016/S1474- 4422(18)30025-5. Epub 2018 Feb 3.
- Messina S, Vita GL. Clinical management of Duchenne muscular dystrophy: the state of the art. Neurol Sci. 2018 Nov;39(11):1837-1845. doi: 10.1007/s10072-018- 3555-3. Epub 2018 Sep 14.
- Pascual Morena C, Martinez-Vizcaino V, Álvarez-Bueno C, Fernández Rodríguez R, Jiménez López E, Torres-Costoso AI, Cavero-Redondo. Effectiveness of pharmacological treatments in Duchenne muscular dystrophy: a protocol for a systematic review and meta-analysis. BMJ Open. 2019 Sep 6;9(9):e029341. doi: 10.1136/bmjopen-2019-029341.
- Neuromuscular Disorders of Infancy, Childhood, and Adolescence , A Clinician’s Approach, Second Edition, Edited by: Basil T. Darras, H. Royden Jones, Jr., Monique M. Ryan, Darryl C. De Vivo, Chapter 15, 16, 17,2014