Резюме
Наследственные моторно-сенсорные нейропатии (НМСН) - обширная группа клинически и генетически гетерогенных заболеваний периферической нервной системы, распространенность которых составляет 1:3000 чел. В настоящее время известно более 50 локусов и открыто более 30 генов, ответственных за развитие НМСН. Представлена современная классификация НМСН. В статье рассмотрены современные методы диагностики наследственных моторно-сенсорных нейропатий (НМСН). Таким образом, на сегодняшний день особенно актуальны вопросы последовательности молекулярно-генетического обследования больных НМСН.
Ключевые слова: наследственные моторно-сенсорные нейропатии, клинические признаки, алгоритм диагностики, клинико-молекулярно-генетический анализ
Наследственные моторно-сенсорные нейропатии (НМСН) - большая группа генетически гетерогенных заболеваний, характеризующихся прогрессирующим поражением периферических нервов. Распространенность НМСН составляет 1 на 3000–3500 человек. Считается, что 70% всех хронических нейропатий являются наследственными [1]. Частота наследственной моторно-сенсорной нейропатии (НМСН) I типа диагностируется в 12,9 случая на 100 000 населения. Существуют также варианты болезней IB и IC, частота которых не установлена [10; 16–18]. НМСН являются самыми
распространенными в рассматриваемой группе заболеваний и имеют наибольшее значение в клинической практике [17,20]. Учитывая тот факт, что на сегодняшний день не разработаны методы патогенетической терапии данной патологии, большое практическое значение приобретает раннее выявление заболевания, позволяющее своевременно проводить комплекс лечебно-реабилитационных мероприятий и профилактику заболеваемости в отягощенных семьях, основанную на медикогенетическом консультировании и пренатальной диагностике [26,27].
Первое описание заболевания было сделано французскими исследователями Charcot-Marie-Tooth в 1886 году, которые обозначили их как невральныеамиотрофии [10,19]. В 1968 году P. Dyck и E. Lambert предложили выделять два основных типа НМСН в зависимости от показателя скорости проведения импульса по срединному нерву и морфологических особенностей поражения миелиновой оболочки. 1 тип НМСН, названный демиелинизирующим или псевдо гипертрофическим, характеризуется снижением скорости проведения импульса и формированием луковицеобразных утолщений в миелиновой оболочке, чередующихся с участками де- и ремиелинизации. 2 тип, названный аксональным, характеризуется нормальными или несколько сниженными скоростями проведения импульса по срединному нерву и отсутствием выраженных изменений миелиновой оболочки. В качестве пороговой величины для разделения НМСН 1 и 2 типов принят показатель скорости проведения импульса по срединному нерву в 38 м/сек. В последние годы на основании проведения клинико-молекулярногенетических корреляций предложено выделение промежуточного типа НМСН, при котором значение скоростей проведения импульса по срединному нерву колеблется от 25 до 45 м/сек у больных из одной и той же семьи. Известны семь генетических вариантов НМСН промежуточного типа (MIM 613641, 606482, 608323, 607791, 615376, 616039, 608340). В настоящее время идентифицировано 30 генов и 50 локусов, ответственных за развитие НМСН [2,21,22]. Достигнутый в последние десятилетия прогресс в определении генетических основ НМСН позволил по-новому рассмотреть их классификацию и патогенез и предложена классификационная структура НМСН, основанная на этиологических различиях [16] . Генетические характеристики различных вариантов НМСН представлены в таблице.В группе НМСН описаны нозологические варианты с аутосомно- доминантным, аутосомно-рецессивным, Х-сцепленным рецессивным и Х- сцепленным доминантным типами наследования.
В зависимости от клиническо-генетической характеристики принято выделять семь типов НМСН. К НМСН I типа относятся пять основных генетических вариантов демиелинизирую- щихполинейропатий с аутосомно-доминантным и Х-сцепленным доминантным типом наследования, и СПИ по срединному нерву ниже 38м/сек. По аутосомно-доминантному типу наследуются IА (MIM 118220), IВ (MIM 118200), IС (MIM 601098) и ID (MIM 607678) типы, а IX тип (MIM 302800) имеет редко встречающийся Х-сцепленный доминантный тип наследования [3-8, 23–25].
29
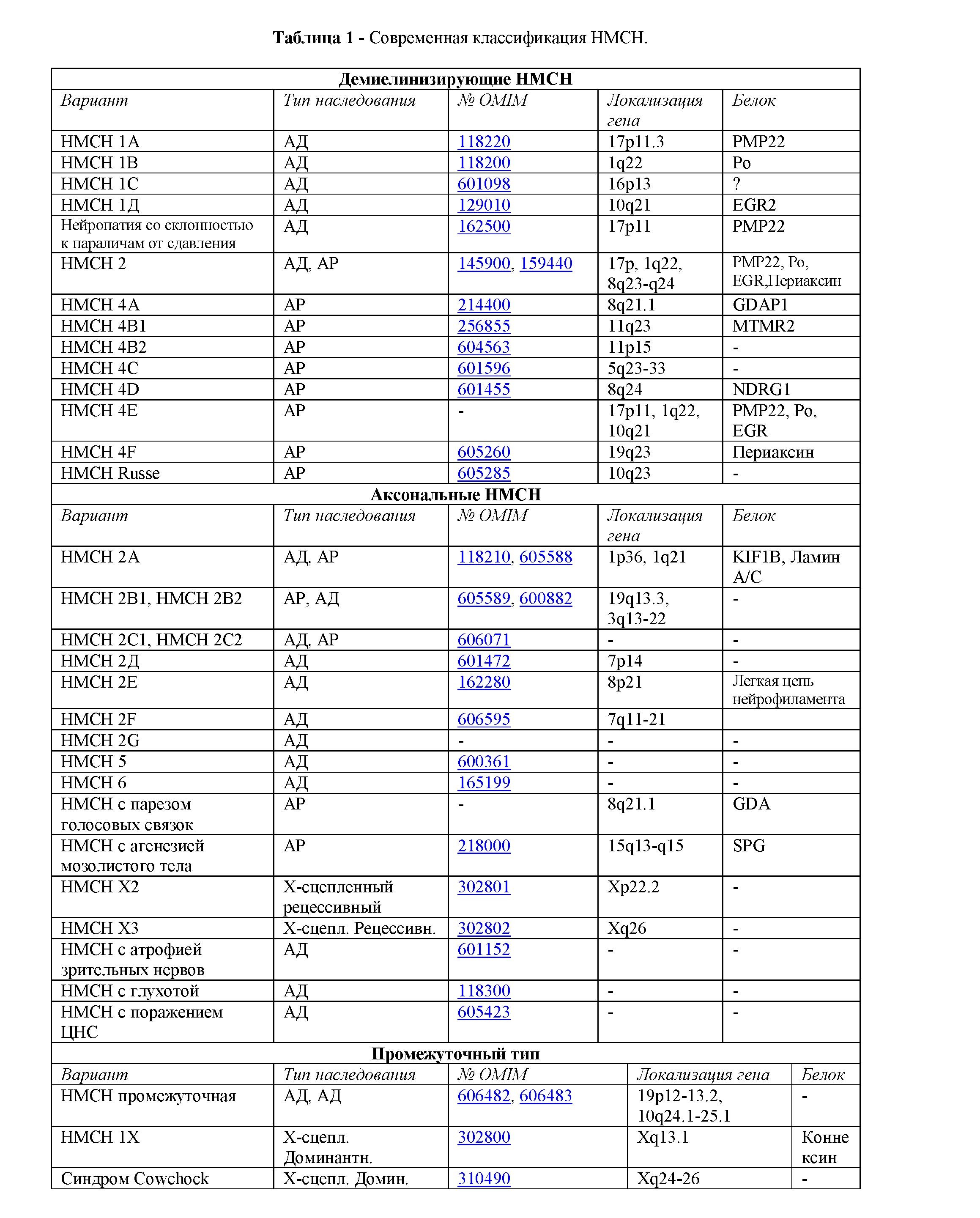
Развитие НМСН I типа обусловлено мутациями в следующих генах: LРМР22 (белок периферического миелина), MPZ (основной белок миелина Р0), GJB1 (белок межклеточных контактов коннексин-32) и EGR2 (ранний фактор транскрипции).
Во втором типе выделено 10 генетических вариантов аксональных полинейропатий с аутосомнодоминантным типом наследования и показателями СПИ по срединному нерву в пределах контрольных значений. Для восьми из этих вариантов идентифицированы гены [2,15]. Третий тип НМСН представлен врожденной демиелинизирующейполинейропатиейДежерина-Сотга. Показано, что это заболевание генетически гетерогенно и его клинические проявления обусловлены мутациями в трех различных генах -РМР22, Р0 и EGR2 . Выделение этого варианта обусловлено особенностями клинических проявлений и аутосомно-доминантным типом наследования [11,13]. Четвертый тип НМСН включает семь генетических вариантов с аутосомно-рецессивным типом наследования, которые относятся как к группе демиелинизирующих, так и аксональных НМСН [9, 12, 14]. Все белковые продукты генов,
ответственных за возникновение НМСН 1 типа, участвуют в формировании миелиновой оболочки периферических нервов, и нарушение их экспрессии приводит к демиелинизации. Эти данные позволили объяснить наличие, характерного для этой группы заболеваний снижения СПИ по срединному нерву. Таким образом, изменение толщины миелиновой оболочки неизбежно приводит к замедлению прохождения импульса и, при проведении электромиографического исследования регистрируется снижение СПИ по периферическим нервам. Изменение объема миелиновой оболочки может быть как в сторону его увеличения, так и в сторону уменьшения. Так, при наиболее распространенном варианте демиелинизирующем варианте НМСН саутосомно-доминантном типе наследования -1А типе (на его долю приходиться не менее 65% все заболеваний этой группы) миелиновая оболочка оказывается утолщенной, что объясняется характером мутации в гене РМР22.
Клинические проявления НМСН: прогрессирующая слабость и гипотрофия мышц стоп, перонеальных мышц голеней, межкостных мышц кистей и сгибательных мышц предплечий, угасание сухожильных рефлексов с мышц верхних и нижних конечностей, появление степпажной походки, расстройства поверхностной и глубокой чувствительности и сенситивно-мозжечковая атаксия. По мере прогрессирования заболевания и нарастания слабости в отдельных мышечных группах возникает деформация кистей и стоп. Характер деформации стоп может быть различным у больных с отдельными вариантами НМСН. Описано возникновение «стопы Фридрейха», полой или плоской стопы. Кисть деформируется по типу «когтистой лапы» или «обезьяньей лапы».
Первыми в патологический процесс вовлекаются мышцы стоп и голеней, в то время как поражение мышц дистальных отделов верхних конечностей возникает спустя несколько месяцев или лет от момента манифестации заболевания. Характерным проявлением НМСН являются расстройства чувствительности в зоне пораженных мышц, характер которых может варьировать при различных генетических вариантах и модифицироваться по мере прогрессирования заболевания. Так, в начальных стадиях некоторых демиелинизирующих полинейропатий может отмечаться гиперестезия стоп и кистей, которая, по мере прогрессирования заболевания, сменяется гипостезией [2,7,9].
Наиболее простой алгоритм диагностики наследственного заболевания состоит из трех последовательных этапов: 1) выявление симптомов и признаков, типичных для клинической картины наследственного заболевания; 2) уточнение диагноза при помощи рутинных методов параклинической диагностики; 3) окончательное подтверждение диагноза в случае обнаружения мутации гена, ответственной за возникновение заболевания. Примером выполнения такого алгоритма может служить диагностика мышечной дистрофии Дюшенна: 1) выявление симптомов и признаков, типичных для мышечной дистрофии Дюшенна, у мальчика в возрасте 3–6 лет; 2) уточнение диагноза на основании выявления высокого (в 10 или более раз превышающего норму) уровня креатинкиназы сыворотки крови; 3) окончательное подтверждение диагноза в случае обнаружения мутации гена дитстрофина (DMD)[28,29]. Однако выполнение такого алгоритма возможно не во всех случаях, поскольку: 1) присущий многим наследственным заболеваниям фенотипический полиморфизм не всегда позволяет клиницистам точно определить нозологическую форму; 2) рутинные методы параклинической диагностики не всегда позволяют уточнить клинический диагноз, так как имеют низкую специфичность и чувствительность; 3) генетическая гетерогенность заболеваний и наличие редких форм значительно усложняют и снижают ценность результатов молекулярно-генетических исследований, направленных на поиск конкретной известной генетикам мутации.
Диагноз подтверждается на основании особенностей клинических проявлений и признаков поражения периферических нервов при проведении ЭМГ исследования. На основании СПИ по срединному нерву проводится отнесение заболевания к демиелинизирующему или аксональному варианту НМСН. Дальнейшая дифференциация происходит на основании типа наследования, возраста манифестации заболевания и особенностей клинических проявлений. При выявлении у пробанда СПИ ниже 38 м/с необходимо в первую очередь исследовать мутации генов белков миелиновой оболочки нерва. В семьях с аутосомно-рецессивным наследованием заболевания последовательность молекулярногенетического исследования генов построена в зависимости от частот встречаемости мутаций в различных генах при аутосомно-рецессивных формах миелинопатий: GDAP1— SH3TC2 — MTMR2 — EGR2. В семьях с аутосомно-доминантным типом наследования и при наличии единственного больного в семье, важным признаком, позволяющим спланировать последовательность исследования мутаций является СПИ по срединному нерву. При её резком снижении (<10 м/с) имеет смысл начинать поиск мутаций с генов MPZ — EGR — LITAF. При СПИ, колеблющихся в промежутке от 10 до 30 м/с наиболее частой причиной заболевания является дупликация на хромосоме 17р11.2-р12 в области гена РМР22, а второй по частоте причиной болезни у мальчиков являются наследуемые Х-сцепленно доминантно мутации гена GJB1. Важными диагностическими признаками, позволяющими спланировать алгоритм ДНК-диагностики, являются тип наследования и возраст манифестации заболевания. Кроме того, есть единственная форма аксонопатии, при которой преимущественно поражаются периферические нервы верхних конечностей — НМСН 2D и причиной которой являются мутации гена GARS. Как известно, единственным надежным способом подтверждения диагноза любого наследственного заболевания является молекулярно-генетическая диагностика, доступная в настоящее время при многих заболеваниях.
Идентификация генетического варианта необходима для решения ряда проблем, основными из которых являются: определение генетического статуса родственников пробанда, определение риска рождения у них больного ребенка и планирование способов дородовой диагностики. Однако, существование генетической гетерогенности и значительного сходства клинических проявлений НМСН создают значительные трудности при проведении такой диагностики с использованием дорогостоящих методов ДНК анализа. Это обусловливает необходимость создания алгоритма идентификации генетического варианта НМСН, который позволит сократить временные и материальные затраты на проведение диагностического этапа и повысит его эффективность. В основу такого алгоритма должны быть положены различия в частоте встречаемости различных вариантов НМСН, возрасте начала, типах наследования, показателях СПИ по срединному нерву и особенностях клинических проявлений и течения заболевания. Таким образом, для планирования алгоритма ДНК диагностики с целью выявления генетического варианта врачу-генетику необходимо: 1) провести генеалогический анализ; 2) определить возраст манифестации заболевания; 3) получить показатели С1Ш по срединному нерву; 4) получить результаты неврологического осмотра. Суммарный анализ этих показателей позволит, поставить диагноз периферической нолинейропатии, определить тип наследования заболевания и отдифференцировать аксональные и демиелинизирующие варианты НМСН.
Выводы. В заключение необходимо отметить, что достаточно высокая распространенность отдельных форм НМСН и часто поздняя диагностика, обуславливают необходимость дальнейшего изучения эпидемиологии НМСН в отдельных территориально-этнических регионах и совершенствования комплекса клинико-генетических исследований, оптимальных для исследуемого региона. Ранняя диагностика НМСН позволит своевременно проводить терапевтические и реабилитационные мероприятия, способствующие замедлению темпов прогрессирования заболевания, а также предупреждение данной патологии в последующих поколениях. Необходимо, на наш взгляд, шире информировать практических врачей, в том числе оказывающих медицинскую помощь в амбулаторнополиклинических условиях, о доступности современных способов верификации диагноза редких наследственных заболеваний нервной системы. Для улучшения качества и своевременности медицинской помощи для таких пациентов целесообразна организация специализированных клиникодиагностических кабинетов.
Литература
- DeJongheP. etal. Molecular diagnostic testing in Charcot-Marie-Tooth disease and related disorders. Approaches and results //Ann N Y AcadSci, 1999. 883: p. 389-96.
- http://www.molgen.ua.ac.be/CMTMutations/Home/Default.cfin.
- Дадали Е.Л., Угаров И.В., Щаркова И.В., Кириленко Н.Б. Проблемы классификации наследственных нейропатий.//Медицинская генетика.- 2003-.No5- С.194-200.
- Bergoffen J., Scherer S.S., Wang S. et al. Connexin mutations in X-linked Charcot-Marie-Tooth disease.// Science.- 1993.- V. 262.- P. 2039-2042.
- De Jonghe P. et al. Molecular diagnostic testing in Charcot-Marie-Tooth disease and related disorders. Approaches and results //Ann N Y AcadSci, 1999. 883: p.389-96.
- De Jonghe P., Timmerman V., Nelis E. et al. Charcot-Marie-Tooth disease and related peripheral neuropathies //J PeripherNervSyst- 1997,- V. 2,- P. 370-387.
- Mersiyanova, l.V. et al. Screening for mutations in the peripheral myelin genes PMP22, MPZ and Cx32 (GJB1) in Russian Charcot-Marie-Tooth neuropathy patients // Hum Mutat, 2000. - V.15.- P. 340-347.
- Roa B.B., Warner L.E., Garcia C.A. et al. Myelin protein zero (MPZ) gene mutations in non duplication type 1 Charcot-Marie-Tooth disease.//Hum Mutat- 1996- V. 7.- P. 36-45.
- Claramunt R, Pedrola L., Sevilla T. et al. Genetics of Charcot-Marie-Tooth disease type 4A: mutations, inheritance, phenotypic variability, and founder effect //J Med Genet- 2005.- V.42.- P. 358-365.
- Вельтищев Ю.Е. Наследственные болезни нервной системы. М., 1998.
- .Mersiyanova, l.V. et al. Screening for mutations in the peripheral myelin genes PMP22, MPZ and Cx32 (GJB1) in Russian Charcot-Marie-Tooth neuropathy patients // Hum Mutat, 2000. - V.15.- P. 340-347.
- Nelis E., S. Erdem, P.Y. Van Den Bergh, et al., Mutations in GDAP1: autosomal recessive CMT with demyelination and axonopathy//Neurology.- 2002.-V.59.- P. 1865-1872.
- Roa B.B., Warner L.E., Garcia C.A. et al. Myelin protein zero (MPZ) gene mutations in non duplication type 1 Charcot-Marie-Tooth disease.//Hum Mutat- 1996- V. 7.- P. 36-45.
- Senderek J., Bergmann C., Weber S. et al. Mutation of the SBF2 gene, encoding a novel member of the myotubularin family, in Charcot-Marie-Tooth neuropathy type 4B2/llpl5.//Hum Mol Genet.- 2003.-V.12.-P. 349-356.
- Zuchner S., Vance J.M. Mechanisms of disease: a molecular genetic update on hereditary axonal neuropathies./'Nat ClinPract Neurol.- 2006.- V.2.- P. 45-53.
- Иллариошкин С.Н., Иванова-Смоленская И.А., Маркова Е.Д. ДНК-диагностика медико-генетическое консультирование в неврологии. М., 2002.
- Dyck P.J., Chance P.F., LeboR., Carney J.A. Hereditary motor and sensory neuropathies // Peripheral neuropathy. 3 ed. Phila-delphia, 1993. P. 1094–1136.
- EmeryA. E.H. Population frequenciesof inherited neuromuscular diseases: a worldsurvey // Neuromusc. Disord. 1991. Vol.103.P.19–29.
- Давиденков С.Н. Наследственные болезни нервной системы. М., 1932.
- Chance P.F., FischbeckK.H. Molecular genetics of Charcot–Mari–Tooth disease and related neuropathies // Hum. Mol. Genet.1994. Vol. 3. P. 1503–1507.
- Murphy SM, Laura M, Fawcett K, et al. Charcot-Marie-Tooth disease: frequency of genetic subtypes and guidelines for genetic testing. Journal of Neurology, Neurosurgery and Psychiatry 2012; 83 (7): 706–710.
- Di Vincenzo C, Elzinga CD, Medeiros AC, et al.The allelic spectrum of Charcot-Marie-Tooth disease in over 17,000 individuals with neuropathy. Molecular Genetics & Genomic Medicine 2014; 2 (6): 522–529.
- Dadali EL, Ugarov IV, Scharkova IV, etal.The problems of classification of hereditary neuropathies. Medical Genetics 2003; 5: 194–200. Russian (ДадалиЕ. Л.,УгаровИ. В., ЩарковаИ. В. идр. Проблемы классификации наследственныхнейропатий. Медицинская генетика 2003; 5: 194–200).
- MersiyanovaIV, IsmailovSM, PolyakovAV, etal. Screening for mutations in the peripheral myelin genes PMP22, MPZ and Cx32 (GJB1) in Russian Charcot-Marie-Tooth neuropathy patients. Hum Mutat 2000; 15: 340–347.
- Taylor RA, Simon EM, Marks HG, et al. The CNS phenotype of X-linked Charcot-Marie-Tooth disease: more than a peripheral problem. Neurology 2003; 61 (11): 1475–1478.
- Левин, О. С. Наследственные моторно-сенсорные невропатии / О. С. Левин. – Полинейропатии. – М.: Мединформ. агентство, 2005. – 496 с.
- Вельтищев, Ю. Е. Наследственные болезни нервной системы / Ю. Е. Вельтищев. – М.: Медицина, 1998. – 496 с.
- Ё.Н. Маджидова, У.Т. Омонова. Метаболическая коррекция двигательных нарушений у детей с прогрессирующими мышечными дистрофиями Дюшенна/Беккера.// Медицинский журнал Узбекистана. – 2018. – №2.– С.60-63.
- Маджидова Ё.Н., Омонова У.Т., Бобоев К.Т. Молекулярно-генетический анализ микросателлитных STR-45 (СА)28, STR-49(СА)24, STR-50 (АС)16 повторы гена dmd у детей с прогрессирующими мышечными дистрофиями Дюшенна и Беккера. //Журнал теоретической и клинической медицины. – 2018. – №2.– С.76-79.