Disorders of oxidative phosphorylation processes in mitochondria are associated with many diseases of the nervous and muscular system, endocrine organs, as well as the heart, kidneys, eyes, which have a severe clinical course and cause disability. Long-term mitochondrial dysfunction leads to insufficient energy supply of cells, disruption of many other important metabolic processes, the further development of cell damage, up to cell death. Coenzyme Q10 is able to affect these processes. Coenzyme Q is necessary for the normal functioning of living organisms and, above all, for the functioning of tissues with a high level of energy metabolism.
Mitochondria are tiny organelles of eukaryotic organisms that produce energy for all biochemical processes. According to the most popular version, mitochondria were once independent units of life, purple photosynthetic bacteria, but in the process of evolution were absorbed by ancient microorganisms, archaea, and began to produce energy for them. Each cell can contain more than a thousand mitochondria, for example, heart muscle cells have up to 5000 mitochondria. Molecular motors, ATP synthases, built into the inner membrane of the mitochondria, under the influence of proton flow rotate and synthesize the main source of energy adenosine triphosphate (ATP). It is estimated that the body of an adult synthesizes and consumes about 40 kg of ATP per day. Mitochondria, unlike other organelles, have their own 2-10 copies of deoxyribonucleic acid (DNA), mitochondrial DNA (mtDNA).
Mitochondria are the main intracellular source of reactive oxygen species (ROS). The corresponding intracellular balance between ROS production and its suppression can be changed due to the intervention of many factors that are beyond the cell control capabilities (exogenous chemical agents, radiation, viral and bacterial infection), and can be programmed by the cell itself (apoptosis and aging), leading to a phenomenon having a common name "oxidative stress". It is believed that mitochondrial nonspecific permeability induction is the cause of redevelopment of the redox potential in the cell and is often used as a model of oxidative stress in vitro [1].
Mitochondria are cellular organelles that perform important functions: supply of cells with energy in the form of ATP, generation and regulation of reactive oxygen species, regulation of calcium ions in the cytoplasm, initiation of apoptosis.
Disorders of these organelles play a leading role in the origin and clinical manifestations of mitochondrial diseases caused by mutations of mitochondrial or nuclear DNA genes encoding energy metabolism. At the same time, it was found that mitochondrial dysfunction and accumulation of mitochondrial mutations in tissues make a significant contribution to the aging process, as well as to the pathogenesis of a number of diseases characterized by neurodegeneration, in particular, Alzheimer's disease. Mutations lead to increased generation of free radicals, reduced levels of ATP and energy deficiency of cells.
Currently, the term "oxidative stress" is used to refer to a wide group of various interrelated phenomena, including increased production of reactive oxygen species and oxidative damage to the molecular components of the cell. The relationship between these phenomena are not fully elucidated, but probably plays an important role in the oxidative damage of mitochondria, autocatalytically leading to increased generation of ROS. The mechanisms of ROS formation by mitochondria under oxidative stress are still unclear. Numerous data obtained in experiments with isolated mitochondria and submitochondrial particles indicate that the main superoxide-forming components of the respiratory chain are NADH: ubiquinonoxide reductase (complex I) and ubiquinone-cytochrome C reductase (complex III). However, it is not clear which component of complex I serves as a single-electron donor for oxygen recovery. Moreover, in physiological conditions, a high level of NADH is maintained in the cells, which may interfere with the formation of superoxide complex. Probably for this reason, experiments on cell cultures give conflicting results on the role of complex I in the generation of ROS. Inhibition of complex I activity in cell culture can lead to both an increase and a decrease in ROS levels depending on the cell type and the stimulus causing oxidative stress. Such ambiguity indicates the complexity of the mechanisms of ROS generation by mitochondria under physiological conditions. The reaction of cells to oxidative stress includes a number of protective mechanisms. First of all, there is activation and additional expression of numerous antioxidant systems, some of which are aimed at protecting mitochondria.
Damaged mitochondria pose a significant danger to cells as both sources of ROS and consumers of NAD(P)H and ATP, so there are mechanisms for the destruction of such organelles. Finally, in the case of exhaustion of all the possibilities of protection, the cell can run a program of death (apoptosis), to avoid the spread of stress in the surrounding tissue. So, earlier it was hypothesized that hydrogen peroxide, formed in the process of cell death, can serve as a signal that causes the death of neighboring cells [29]. This signal transmission can be one of the mechanisms to protect the body from infection by pathogens. For example, with a localized viral infection, a zone of dead cells may occur around the zone of virus reproduction, preventing the spread of infection throughout the body. In addition, apoptosis signal transmission plays an important role in tumor therapy. Induction of apoptosis in the tumor under the action of cytotoxic chemotherapy or radiation can affect only part of the cells, and the death of the rest can occur under the action of hydrogen peroxide. In this case, tumor cells protected from apoptosis caused by cytotoxic drugs (for example, as a result of mutations in signal pathways dependent on p53 proteins) may be destroyed, while apoptosis induced by H2O2 does not require signal transmission by this way. The main mechanism of cell death in this case is probably associated with oxidative damage of mitochondria under the action of external H2C >2, which stimulates the production of "secondary" ROS by mitochondria and thus causes cell apoptosis. Research in this area is of great interest due to the fact that oxidative stress, accompanied by damage to the mitochondria, is observed in many serious pathologies, which are an urgent problem for health care, in particular with heart attacks, strokes, neurodegenerative diseases.
Elucidation of the role of mitochondrial bioenergetic functions disturbance in the induction of oxidative stress, as well as study of the mechanisms of cell protection from increased ROS production by mitochondria will allow to fight these diseases more effectively [2].
Human mitochondrial DNA is a double-stranded ring molecule containing 16,600 base pairs. It encodes 22 molecules of mRNA, 2 rRNA and 13 polypeptides of the respiratory chain enzymes. Thus, human mitochondria, like other eukaryotic organisms, have their own genetic system, which involves mtDNA, mitochondrial ribosomes, tRNA and proteins, providing the processes of transcription, translation and replication of mtDNA [3, 4]. The processes of oxidative phosphorylation in mitochondria provide the formation of the main energy substrate of all metabolic processes (ATP), because mutations of structural genes mtDNA, often lead to various violations of energy metabolism in cells. These disorders are the basis of pathological conditions called human mitochondrial diseases [5, 6]. In addition, mtDNA is transmitted exclusively with the cytoplasm of the egg, since sperm do not contain mitochondria. Because mitochondrial genes and mitochondrial diseases are inherited only through the maternal line. The first information about mitochondrial diseases appeared in 1962, when a young woman was determined to have a hypermetabolic state with structurally abnormal mitochondria and a violation of the respiratory chain organelles symptoms, known today as Lufta disease. Further molecular mechanisms of mitochondrial diseases were studied using polymerase chain reaction, Southern DNA hybridization and other molecular genetic methods [7, 8, 9]. And to date, more than 200 mitochondrial diseases are known, which are based on violations of the oxidative phosphorylation system. There are two main groups of mutations occurring in mtDNA: 1) mutations associated with the deletions of more or less significant fragments of the molecule (approx.: progressive external ophthalmoplegia, Pearson's syndrome, manifested by dysfunction of the bone marrow, pancreas and other pathological conditions); 2) point mutations-in this case there is a mitochondrial polymorphism, when in each cell there is both normal and mutant mtDNA. With subsequent replication of mtDNA and cell divisions formed variants of cells, accumulating normal and mutant molecules in different ratios. This or that polymorphism can be inherited by all children from the mother [10,11]. Examples include Leber optical neuropathy associated with progressive optic nerve atrophy leading to blindness before the age of 20, melas syndrome manifested in encephalopathy (headaches, convulsions, impaired mental development), and other diseases. Attention is drawn to the fact that the effect of mutations in mtDNA on the function of a tissue depends on the level of its consumption of ATP. Since the most energydependent are the Central nervous system and the muscular system, clinically such mutations are manifested most often in the form of various neuropathies and myopathies, affects the heart, kidneys, eyes, endocrine organs. In most cases, these diseases are characterized by a large variability of existing symptoms [12, 13, 14]. There is also reason to believe that as the accumulation of mutations of mtDNA in the somatic cells of the individual in them is a progressive process of imbalance of oxidative phosphorylation, which is one of the main causes of aging. In the elderly, mutations of mtDNA are detected with a frequency of 50%. It is interesting to note that mutations can occur not only in the mitochondrial genome, but also in the nuclear one. However, mutations in mtDNA accumulate 10-20 times faster than in nuclear DNA. This is due to the fact that mtDNA is not protected by histone proteins, and the ancient, inherited from bacterial ancestors, the mechanisms of repair of its damage are imperfect. The spectrum of mitochondrial diseases is extensive, disorders of energy metabolism in cells are the basis of the following syndromes: Alzheimer's disease; Parkinson's disease; gastrointestinal reflux; insulin-independent diabetes mellitus; lethal pediatric mitochondrial myopathy; maternal inherited Leia syndrome; mitochondrial myopathy; dementia and chorea syndrome; myoclonic epilexy syndrome; • diabetes mellitus and hearing loss; myopathy and diabetes syndrome; Multisystem mitochondrial disorder syndrome; sudden infant death syndrome;lactic acidosis and stroke-like episodes; progressive myoclonic epilepsy syndrome; family bilateral necrosis of striated bodies; ataxia, myoclonia and hearing loss syndrome.
Especially a large number of hereditary human pathology, including mitochondrial diseases, is observed in the practice of pediatricians [16]. It is known that at least a third of all children with disabilities in the symptom complex of their diseases have signs of polysystem disorders of cellular energy. The presence of mitochondrial disorders in such pathologies in children was revealed: connective tissue diseases (Marfan and Ehlers-Danlos syndromes), tuberous sclerosis, a number of non-endocrine syndromes accompanied by growth retardation (osteochondrodysplasia, silver-Russell syndrome, etc.), the influence of mitochondrial insufficiency on the course of a number of cardiac, surgical and other diseases, including diabetes mellitus.
As a result, long-term mitochondrial dysfunction leads to insufficient energy supply to cells, disruption of many other important metabolic processes, further development of cell damage, up to cell death [17]. The drugs that can affect these processes, to improve the energy cells are coenzyme Q10 (ubiquinone), L-carnitine, b vitamins, etc. Ubiquinone is a substance of endogenous nature, a mandatory component of the membranes of mitochondria, lysosomes, Golgi apparatus, plasma membrane [18]. It was first isolated in 1957 from the bull's heart by a group of American researchers led by Frederick crane. A year later, Professor faulker's team, supported by the pharmaceutical company Merck, Sharp and Dohme, identified the chemical structure and function of the newly discovered substance. Professor faulker devoted his entire life to the study of Coenzyme Q 10. For 40 years, his group has studied the properties of Coenzyme Q10 with the aim of determining the possibility of the use of Coenzyme Q10 in medicine. The first successful experience of the drug in CHF was noted in 1967. Since then, every year the number of sections of medicine in which the use of Coenzyme Q 10 showed favorable clinical effects increases [19].
Coenzyme Q10 in mitochondria is involved in the synthesis of ATP as an electron carrier, which matches the processes of electronic transport and oxidative phosphorylation. Coenzyme Q10 is a necessary link for the transfer of electrons from complexes I and II to the complex III of the respiratory chain. With its lack (difficulty in electron transfer through the respiratory chain), complexes I and III become the main generators of superoxide anion-radicals of oxygen [20-22]. Coenzyme Q10 takes part not only in the energy metabolism of the cell, but also occupies a Central place in its antioxidant defense. The development of many diseases is accompanied by oxidative tissue damage due to the generation of active forms of oxygen in them. During intensely flowing redox processes in cells form endogenous free radicals such as superoxide, nitric oxide, hydrogen peroxide, lipid radicals and other reactive oxygen species damage the cell membrane and collagen fibers, capable of destroying the free amino acids (cysteine, lysine) [23]. To prevent the harmful effects of reactive oxygen species, cells of the body use antioxidant systems, low molecular weight and specialized antioxidant enzymes-superoxide dismutase, catalase, glutathione peroxidase, glutathione (Tripeptide). However, with the development of processes leading to the activation of oxidative metabolism, the reserve of antioxidant factors is not enough, so it is necessary to receive such substances as vitamins E and C, provitamin A, plant polyphenols (flavonoids) and quinones of animal origin (vitamin K, coenzyme Q10), from the outside, with food. A feature of coenzyme Q10 is the ability to regenerate, that is, to restore its oxidized form with the help of enzyme systems of the body and antioxidants of non-enzymatic nature (ascorbate, tocopherol), which returns its antioxidant activity. Coenzyme Q10 is produced in the human body constantly, since birth, but after 30 years of its reproduction is steadily declining, and by 70-80 years reaches the same level as in infancy. In addition, the need for coenzyme Q10 increases with active physical and mental stress, as well as diseases [24], leading to the development of energy-deficient conditions. These conditions are very different and include a number of diseases. The main causes which may lead to deficiency of CoQ10 in humans: reduced biosynthesis, and increased consumption by the body. The main source of coenzyme is biosynthesis, it requires correct work of at least 12 genes, and mutation in them can lead to deficiency. Also, Q10 deficiency can develop due to other genetic defects, in particular due to mutation of mitochondrial DNA, genes ETFDH, APTX, FXN, and BRAF.
Table 1. Coenzyme Q content in various products
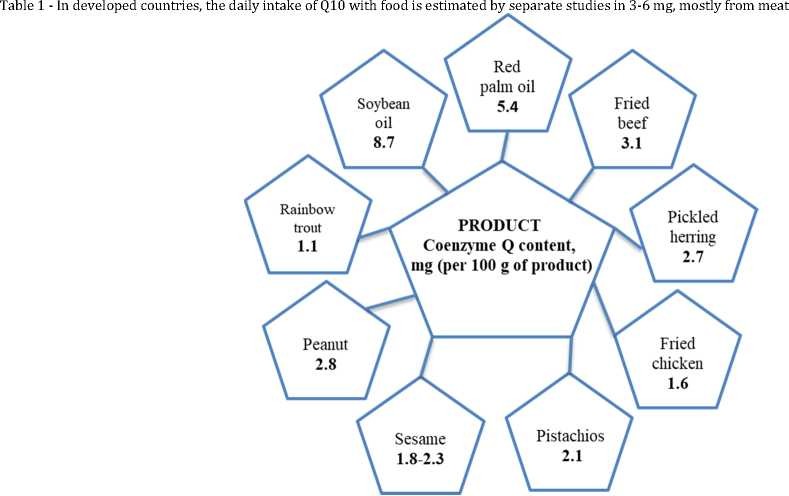
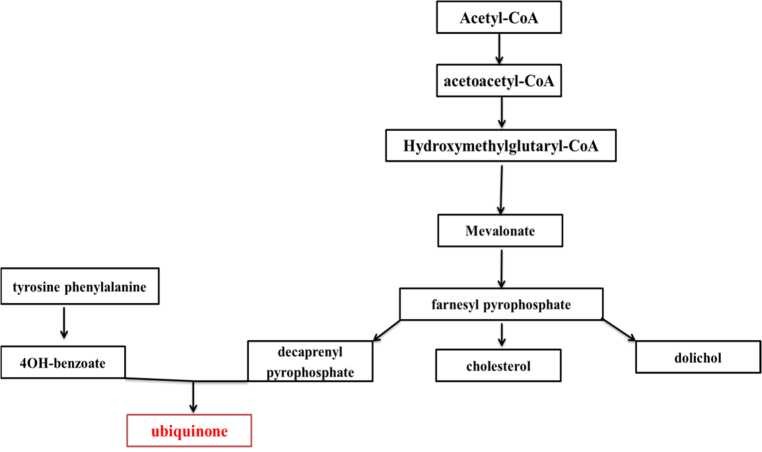
In humans, coenzyme Q10 is synthesized in all cells. Synthesis is carried out from mevalonic acid, and products of tyrosine and phenylalanine metabolism with the participation of vitamins B2, B3, B6, B12, C, folic and Pantothenic acids, as well as a number of trace elements. It is a complex, multistage process regulated by several enzyme systems [25]. With a deficiency of vitamins and minerals, violations of the regulatory enzyme systems and even outside of any pathology endogenous biosynthesis Q10 does not meet the needs of the body.
Figure 1 - A simplified scheme of the biosynthesis of ubiquinone in the body
As one of the links of the respiratory chain, ubiquinone transfers electrons from NADPH-dehydrogenase complex (complex I) and succinate dehydrogenase complex (II) to complex III, and thus participates in the synthesis of macroergic molecules ATP. Q10 is absolutely necessary and indispensable for oxygen-dependent energy formation. In the absence of Q10, this process is interrupted.
Tens of clinical trials involving thousands of patients with various forms of cardiovascular disease have been conducted. Most patients took coenzyme Q as part of complex therapy immediately after diagnosis. In some patients, the improvement was striking: the heart regained normal size and contractile function. In the more advanced forms of the disease there was no complete recovery, but there was a clear improvement.
In addition, the need for coenzyme Q10 increases with active physical and mental stress, as well as with diseases [26], leading to the development of energy-deficient conditions. These conditions are very different and include a number of diseases. The initial pathogenetic link of these diseases is a mutation in the mitochondrial genome. The primary mitochondrial diseases currently include the group of encephalomyopathies: MERRF syndrome (myoclonusepilepsia, "torn red fibers"), MELAS syndrome (mitochondrial encephalopathy, lactate acidosis, stroke-like episodes), Kearns-seyr syndrome (pigment retinitis, external ophthalmoplegia, heart blockade, ptosis, cerebellar syndrome), Pearson's syndrome (bone marrow damage, pancreatic and hepatic dysfunction), Leber's disease and a number of OTHER, not always clearly defined, conditions. These diseases have a number of similar signs with systemic mitochondrial insufficiency (muscle weakness, ptosis, ophthalmoplegia, mental retardation and biochemical changes such as increased levels of pyruvate and lactate in the blood) [27]. Hereditary mitochondrial defects associated with nuclear genome damage have been less studied. At present, relatively few of them are known (various forms of infant myopathies, Alpers', Leia's, Bart's, Menkes' diseases, carnitine insufficiency syndromes, some enzymes of the Krebs cycle and mitochondrial respiratory chain) [28]. Complex energotropic therapy of these diseases, including coenzyme Q10, cytochrome C, L-carnitine and some others, allows to achieve a significant clinical effect. The result of treatment is an increase in body weight, a decrease in the severity of cardiovascular disorders, a decrease in the frequency of vomiting, seizures, a decrease in the severity of manifestations of encephalopathy and myopathy, a decrease in fatigue depending on the scope of manifestations of the pathological process.
Treatment of mitochondrial diseases is carried out in two main directions. The first is to increase the efficiency of energy metabolism in tissues. To do this, drugs are introduced whose components provide tissue respiration and oxidative phosphorylation (thiamine, Riboflavin, nicotinamide, coenzyme Q10, vitamin C, cytochrome, etc.). The second direction of therapy is prevention of mitochondrial membrane damage by free radicals with the help of antioxidants (vitamins E and C) and membrane protectors. In conclusion, it should be emphasized that the discovery in recent years of the role of mitochondria in drug sensitivity, their key role in aging, apoptosis and neurodegenerative disorders has led to the creation of mitochondrial medicine.
REFERENCES
- 1 Turnbull H. E., Lax N. Z., Diodato D., Ansorge O., Turnbull D. M., The mitochondrial brain: From mitochondrial genome to neurodegeneration // Biochim. Biophys. Acta. 2010. V.1802. Р. 111-121.
- 2 Mazunin I. O., Volodko N. In. Starikovskaya E. B., Sukernik R. I. Mitochondrial genome and human mitochondrial diseases // Mol. Biol. 2010. Vol. 44., №5. Р. 755 772.
- Illarioshkin S. N. Algorithm of diagnostics of mitochondrial encephalomyopathies // Atmosphere. Nervous disease. 2007. Vol.3. P. 2327.
- Haas R. H., Parikh S., Falk M. J., Saneto R. P., Wolf N. I., Darik N., Wong L. J., Cohen B. H., Naviaux R. K., The in-depth evaluation of the suspected mitochondrial disease // Mol. Genet. Metab. 2008. V. 94. Р. 16-37.
- Zakharova I. N., Obynochnaya E. G., Skorobogatova E. V., Malashina O. A. The influence of antioxidant on the basis of the activity of lipid peroxidation and antioxidant protection in pyelonephritis in children // Pediatrics. 2005. № 4. Р. 48-54.
- Klyuchnikov S. O., Gnetneva E. S. Ubiquinone. Theory and clinical practice // Pediatrics. Speransky. 2008. №3. Р. 169-175.
- Crane F.L. Biochemical functions of coenzyme Q10 // Journal Coll. Nutr. 2001. №20. Р. 591-598.
- Hsu C.H., Cui Z., Mumper R.J., Jay M. Preparation and Characterization of Novel Coenzyme Q10 // Nanoparticles Engineered from Microemulsion Precursors. 2003. №32. Р. 269-280.
- Joshi S.S., Sawant S.V., Shedge A., Halpner A.D. Comparative bioavailability of two novel coenzyme Q10 preparations in humans // Int. Journal Clin. Pharmacol. Ther. 2003. №41(1). Р. 42-48.
- Singh R.B., Niaz M.A., Rastogi S.S. et al. Effect of hydrosoluble coenzyme Q10 on blood pressures and insulin resistance in hypertensive patients with coronary artery disease // Journal of Human Hypertension. 1999. V.13., №3. P. 203-208.
- Shults C.W., Beal M.F., Song D., Fontaine D. Pilot trial of high dosages of coenzyme Q10 in patients with Parkinson's disease // Exp. Neurol. 2004. №188. Р. 491-494.
- Soongswang J., Sangtawesin C., Durongpisitkul K., Laohaprasitiporn D., Nana A., Punlee K., Kangkagate C. The effect of coenzyme Q10 on idiopathic chronic dilated cardiomyopathy in children. // Pediatric Cardiology. 2005. №4. P. 361-366.
- Morisco C, Nappi A, Argenziano L et al. Noninvasive evaluation of cardiac hemodynamic during exercise in patients with chronic heart failure: effects of short-term coenzyme Q10 treatment // Molecular Aspects of Medicine. 1994. №15. P. 155-163.
- Mortensen SA, Vadhanavikit s, Defence of coenzyme Q10 in myocardial failure // Drugs under Experimental and Clinical Research. 1984. № 7. P. 497-502.
- Fujita T, Tanayama S, Shirakawa Y. Metabolic fate of ubiquinone-7. I. Absorption, excretion and tissue distribution in rats // Journal Biochem. 1971. №69. Р. 53-61.
- Finsterer J. Treatment of mitochondrial disorders // Eur. J. Paediatr. Neurol. 2010. V.29. Р. 446-452.
- Mazunin I. O., Volodko N. In. Mitochondria: life in a cell and its consequences // Nature. 2010. №10. Р. 3-14.
- Poulton J., Turnbull D. M. 74th ENMC international workshop: mitochondrial diseases // Ehe Netherlands. Disord. 2000. V.10. Р. 460462.
- Vorsanova S. G., Yurov V. N. Medical cytogenetics. M.: Medical Practice-M, 2006. 300 р.
- Coskun P. E., Busciglio J. Oxidative Stress and Mitochondrial Dysfunction in Down's Syndrome: Relevance to Aging and Dementia // Curr. Gerontol Geriatr. Res. 2012. №4. Р. 383-391.
- Wallace D. C, Fan W, Procaccio V. Mitochondrial energetics and therapeutics // Ann Rev. Pathol. 2010. №5. Р. 297-348.
- Venditti P., DiStefano L., DiMeo S. Mitochondrial metabolism Of reactive oxygen species // J. Mitochondrion. 2013. №13. Р. 71-82.
- N. Howell, J. L. Elson, P. F. Chinnery, D. M. Turnbull mtDNA mutations and common neurodegenerative disorders // Trends in Genetics. 2005. №21(11). Р. 583-586.
- Coskun P., Wyrembak J., Schriner S. et al. A mitochondrial etiology of Alzheimer and Parkinson disease // Biochim. Biophys. Acta. 2012. №1820. Р. 553-564.
- Molyneux S. L., Yong et al. Coenzyme Q10: is there a clinical role and a case for measurement? // Clin. Biochem. Rev. 2008. №29. Р. 71-82.
- Ishii N., Senoo-Matsuda N., Miyake K. et al. Coenzyme Q10 can prolong C. elegans lifespan by lowering oxidative stress.// Mech. Aging Dev. 2004; 125: 1: 41-46.
- Littarru G. P., Tiano L. Bioenergetic and antioxidant properties of coenzyme Q10: recent developments // Mol Biotechnol. 2007. №37(1).Р. 31-37.
- Lenaz G., Daurelio M., Merlo Pich M. et al. Mitochondrial bioenergetics in aging // Biochim. Biophys. Acta. 2000. №1459. Р. 397-404.
- Skulachev V.P. Possible role of reactive oxygen species in protection against viral infections // Biochemistry. 1998. vol. 63. Р. 16911694.