Исследована возможность использования масс-спектров как дескрипторов молекулярной структуры при прогнозировании температуры кипения некоторых алкенов. Качество прогнозирования, для набора из 116 соединений, характеризуется коэффициентом корреляции 0.9994 и стандартным отклонением 1.70 К.
Развитие методов прогнозирования физико-химических свойств органических соединений путем установления их корреляций с дескрипторами структуры молекул (QSPR- исследования) является одним из приоритетных направлений современной химии. Прогнозирование позволяет заменить экспериментальное изучение расчетом, а также оценить целевые свойства еще не синтезированных веществ. Вследствие технологической значимости температуры кипения (ТК) вещества и наличия обширных экспериментальных данных, необходимых для формирования обучающих выборок, именно это свойство чаще всего прогнозируется и моделируется. В литературе описано множество QSPR - моделей как для наборов веществ отдельных классов соединений, так и для смешанных наборов различных классов. В то же время прогнозированию ТК алкенов (олефинов) - основных исходных реагентов в химической промышленности - посвящено всего несколько статей. Основной причиной такого положения, на наш взгляд, является наличие диастереомерии у алкенов, значительно осложняющее прогнозирование. Наряду с изомерией углеродного скелета и различием в положении двойной связи возможна геометрическая (цис-, транс-) изомерия, обусловленная большим барьером вращения вокруг двойной связи. Обычно применяемые в качестве дескрипторов структуры топологические индексы, как следует из самого определения, не отражают стереохимической информации и вынуждают применять дополнительно дескрипторы иной природы.
В данной работе представлены результаты использования масс- спектров низкого разрешения в качестве дескрипторов молекулярной структуры алкенов для прогнозирования ТК. Ранее масс-спектры были успешно применены нами при прогнозировании ТК алканов [1] и циклоалканов [2]. Использованы масс- спектры и значения температур кипения 116 алкенов с числом атомов углерода от 3 до 11, имеющиеся на сервере Национального Института Стандартов (МБ^США) [3]. В наборе масс-спектров использованных веществ имеется 123 значения отличных от нуля интенсивностей пиков в диапазоне отношений m/z от 1 до 156. Для получения рабочих значений дескрипторов масс-спектры перед расчетами были преобразованы по формуле
d -
∑ h,t к-1
где d - рабочие значения дескрипторов, используемые в расчетах; n- количество пиков; h - их относительные интенсивности; i – номер вещества; j- номер пика с соответствующим m/z - отношением массы к заряду k-го иона в молекуле с номером i. Элементы dιj формируют матрицу дескрипторов D . Тренировочная выборка состояла из 95, а контрольная - из 21 вещества. Расчеты выполнены с помощью разработанной нами компьютерной программы PROGROC (PROGgramRObustness Calculation). Качество прогнозирования характеризовалось коэффициентом корреляции R между прогнозируемыми и экспериментальными значениями ТК и стандартным отклонением s.
Результаты прогнозирования температуры кипения алкенов приведены в таблицах 1, 2 и на рисунках 1, 2.
Таблица 1 -Экспериментальные [3] и рассчитанные по масс-спектрам значения температуры кипения алкенов (К), контрольная выборка
|
№ |
Соединение |
Экспер. |
Рассч |
Разн |
|
1 |
Г ептен-3 |
369.0 |
369.2 |
-0.2 |
|
2 |
2-Метилпентен-2 |
340.2 |
338.2 |
2.0 |
|
3 |
Цис-2-бутен |
276.9 |
279.7 |
-2.8 |
|
4 |
Транс-нонен-3 |
420.7 |
425.0 |
-4.3 |
|
5 |
3 -Метилгексен-1 |
358.0 |
359.1 |
-1.1 |
|
6 |
2,3,3-Триметилбутен-1 |
351.1 |
350.2 |
0.9 |
|
7 |
Транс-октен-2 |
398.2 |
399.9 |
-1.7 |
|
8 |
Г ептен-2 (цис,транс) |
371.4 |
368.2 |
3.2 |
|
9 |
Цис-3 -метилгексен-3 |
368.6 |
368.3 |
0.3 |
|
10 |
Транс-3-гексен |
340.2 |
337.5 |
2.7 |
|
11 |
2,4-Диметилпентен-1 |
354.8 |
354.7 |
0.1 |
|
12 |
Т ранс-4-ундецен |
466.2 |
467.2 |
-1.0 |
|
13 |
Гексен-1 |
336.6 |
338.3 |
-1.7 |
|
14 |
Октен-2 (цис,транс) |
398.0 |
398.6 |
-0.6 |
|
15 |
Транс-пентен-2 |
309.5 |
311.7 |
-2.2 |
|
16 |
Цис-4,4-диметилпентен-2 |
353.6 |
356.5 |
-2.9 |
|
17 |
3-Этилпентен-1 |
357.3 |
358.8 |
-1.5 |
|
18 |
3,3 - Диметилбутен-1 |
314.5 |
313.7 |
0.8 |
|
19 |
2,4,4-Триметилпентен-2 |
378.0 |
377.3 |
0.7 |
|
20 |
Цис-3 -метилпентен-2 |
340.9 |
342.0 |
-1.1 |
|
21 |
Т ранс-2-ундецен |
465.7 |
467.0 |
-1.3 |
Полученным результатам соответствует ранг матрицы дескрипторов p(D)=78.
149
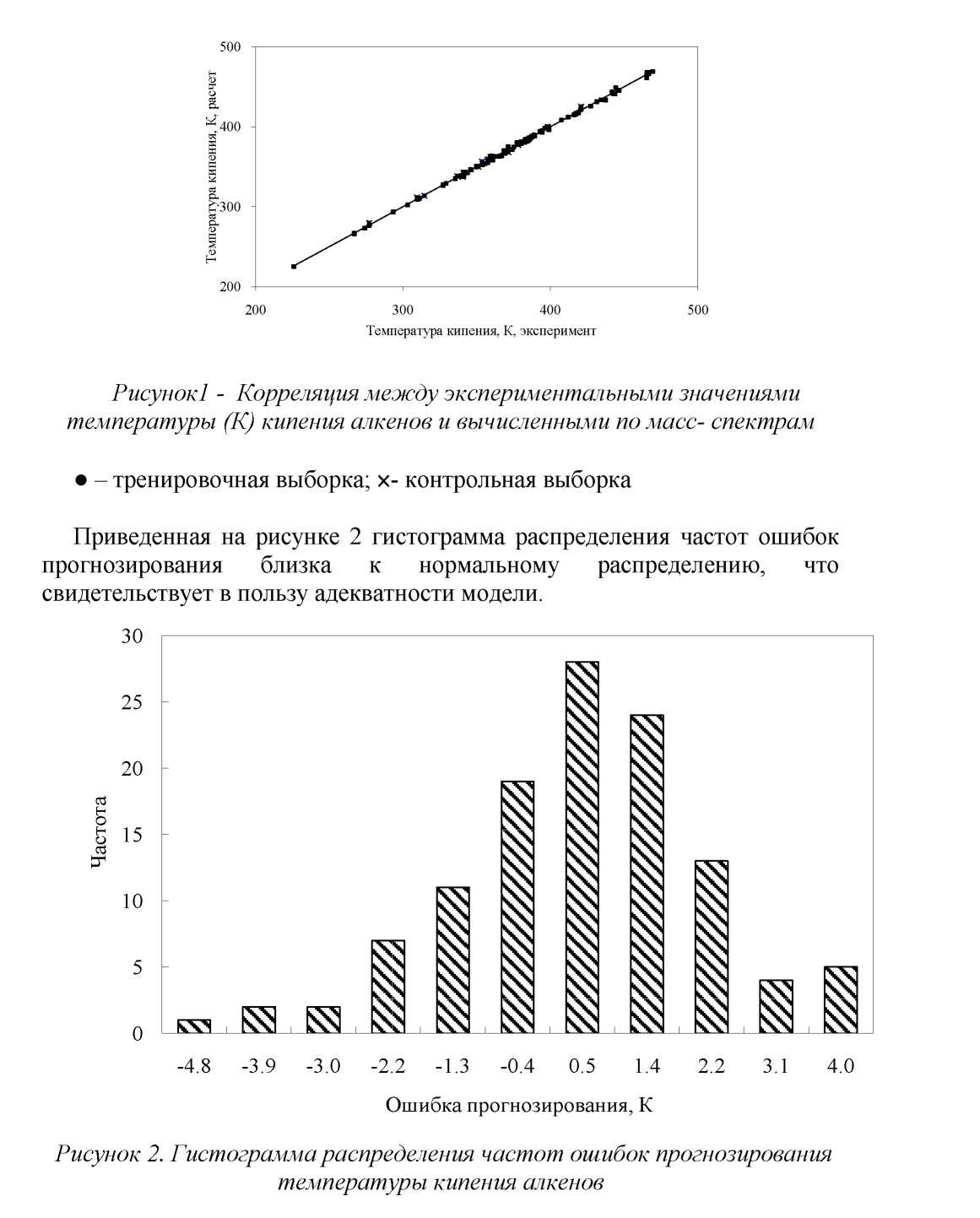
Таблица 2 -Показатели корреляции между экспериментальными и вычисленными значениями температуры кипения алкенов
|
Показатели корреляции |
Весь набор |
Тренировочная выборка |
Контрольная выборка |
|
R |
0.9994 |
0.9994 |
0.9992 |
|
s |
1.70 |
1.64 |
1.88 |
|
Абс. средняя ошибка |
1.29 |
1.23 |
1.59 |
Для оценки качества выполненного нами прогнозирования, обратимся к литературным данным, приведенным в табл.3.
Таблица 3 -Результаты прогнозирования ТК по литературным данным
|
Вещества |
Кол- во |
R |
s |
Л ит. |
|
Алкены, диены |
144 |
0.9998 5 |
0.9 2 |
[4^ ] |
|
Алканы, алкены, диены, алкины |
327 |
0.9998 5 |
1.1 5 |
[4 ] |
|
Алканы |
211 |
0.9991 |
6.0 46 |
[5 ] |
|
Алканы |
7-1 |
0.9968 |
3.8 48 |
[6 ] |
|
Алканы |
160 |
0.9987 |
3.0 6 |
[7 ] |
|
Алканы |
īī¯¯ |
0.9968 |
3.8 48 |
[8 ] |
|
Алканы |
īī¯¯ |
0.9986 |
4.1 87 |
[9 ] |
|
Углеводороды |
296 |
0.997 |
6.3 |
[1 0] |
|
Алканы, спирты |
328 |
0.9990 |
6.0 21 |
[5 ] |
|
Алканы, спирты |
245 |
0.97 |
8.0 |
[1 1] |
|
Набор разных классов |
612 |
0.9823 |
Is. 5 |
[1 2] |
|
Набор разных классов. |
298 |
0.990 |
10. 62 |
[1 3] |
Рассмотрим более подробно результаты работы [4], поскольку они заметно выделяются среди прочих, тем более, что в ней приводятся данныепо прогнозированию ІК алкенов. Авторы этой работы кроме пяти топологических индексов связности использовали в качестве дополнительных дескрипторов дипольный момент и относительную молекулярную массу. Для вычислений использовано предложенное авторами некоторое расширение нейросетевого подхода к QSPR, называемое fuzzy ARTMAP. Следует отметить, что при выделении из общего результата подкласса алкенов (97 веществ), значение R понизится до 0.9982. В этой же статье упоминается о работе Занга и др.[14], в которой для нейросетевого моделировании ІК 85 алкенов приведено значение средней абсолютной ошибки 2 К, максимальная ошибка- 5.7 К.
Сравнивая данные табл. 2 с литературными данными, можно видеть, что полученные нами результаты сопоставимы с лучшими примерами прогнозирования других авторов. Развиваемый нами подход позволяет прогнозировать свойства неизвестных соединений, используя лишь их масс-спектры.
ЛИТЕРАТУРА
- Важев В.В. Прогнозирование температуры кипения алканов по их ИК- и масс- спектрам// Нефть и газ: Алматы. - 2003. - №4. - C. 74-82.
- Важев В.В. Прогнозирование температуры кипения циклоалканов по их масс- спектрам// Нефть и газ: Алматы. - 2004. - №2. - С. 74-80.
- NIST Chemistry WebBook. NIST Standard Reference Database Number 69 - November 1998 Release. http://webbook.nist.gov/chemistry/
- Espinosa G., Yaffe D., Cohen Y., Arenas A., Giralt F. Neural Network Based Quantitative Structural Property Relations (QSPRs) for Predicting Boiling Points of Aliphatic Hydrocarbons // J. Chem. Inf. Comput. Sci. - 2000. - V. 40. - №3. - P. 859-879.
- Cao C., Liu S., Li Z. On Molecular Polarizability: 2. Relationship to the Boiling Point of Alkanes and Alcohols//J. Chem. Inf. Comput. Sci. - 1999. - V. 39.- №6. - P. 1105-1111.
- Ren B. Application of novel atom-type AI topological indices to QSPR studies of alkanes //Computers and Chemistry. - 2002. - V. 26. - P. 357-369.
- Cao C., Jiang L., Yuan H. Eigenvalues of the Bond Adjacency Matrix Extended to Application in Physicochemical Properties of Alkanes//Internet Electron. J. Mol. Des. - 2003. - № 2. – P. 621-641.
- Ren B. Application of novel atom-type AI topological indices to QSPR studies of alkanes //Computers and Chemistry. - 2002. - V. 26. - P. 357-369.
- Krenkel G., Castro E.A., Toropov A.A. 3D and 4D molecular models derived from the ideal symmetry method: prediction of alkanes normal boiling points //Chemical Physics Letters. - 2002. - V. 355. - № 5-6. - P. 517-528.
- Wessel M.D., Jurs P.C. Prediction of Normal Boiling Points of Hydrocarbons from Molecular Structure. //J. Chem. Inf. Comput. Sci. - 1995. - 35. - P. 68-76.
- Hall L.H., Kier L.B. Electrotopological State Indices for Atom Types: A Novel Combination of Electronic, Topological, and Valence State Information. //J. Chem. Inf. Comput. Sci. - 1995. - 35. - P. 1039-1045.
- Katritzky A.R., Lobanov V.S., Karelson M. Normal Boiling Points for Organic Compounds: Correlation and Prediction by a Quantitative StructureProperty Relationship //J. Chem. Inf. Comput. Sci. -1998. - V. 38. - №1. - P. 2841.
- Goll E.S., Jurs PC. Prediction of the Normal Boiling Points of Organic Compounds from Molecular Structures with a Computational Neural Network Model // J. Chem. Inf. Comput. Sci. -1999. - V. 39. - № 6. - P. 974-983.
- Zhang R., Kiu S., Liu M., Hu Z. Neural Networks-Molecular Descriptors Approach to the Prediction of Properties of Alkenes. //Computers Chem. - 1997. - V. 21. - № 5. - P. 335-342.