Резюме
В статье представлена информация о генетической патологии при синдроме-ЦАДАСИЛ. Особое внимание уделено проблеме дифференциальной диагностики ЦАДАСИЛ-синдрома с демиелинизирующими заболеваниями, протекающими со сходной неврологической симптоматикой и нейровизуализационными признаками. Своевременная диагностика синдрома-ЦАДАСИЛ до сих пор остается сложной в клинической практике. Однако знание патогенеза, клиники и тонкостей дифференциальной диагностики позволяет своевременно его диагностировать и провести соответствующее лечение. Приводится описание больного с ЦАДАСИЛ-синдромом. Представлены данные анамнеза, дебюта заболевания, характера течения и результаты лабораторных и инструментальных исследований. Проведена дифференциальная диагностика, обсуждены диагностические ошибки и сделано заключение по диагнозу..
Ключевые слова: синдром ЦАДАСИЛ, дифференциальная диагностика, нейровизуализационные признаки, генетическое исследование.
Введение. Синдром Цадасил является наиболее распространенной формой наследственного поражения сосудов мозга. Патология впервые описана более 30 лет назад в шведской семье, а детально - группой французских исследователей в начале 1990-х годов. Большинство литературных источников представляют собой ретроспективные обзоры случаев из практики или литературные обзоры.
Впервые синдром Цадасил был описан более 30 лет назад в шведской семье, а детально - группой французских исследователей в начале 1990-х годов. Распространенность Цадасил в различных популяциях составляет не менее 1 случая на 100 000 населения, однако предполагается, что реальные цифры существенно выше, поскольку многие случаи Цадасил все еще остаются не диагностированными. Тип наследования синдрома Цадасил является аутосомно-доминантный, одна ненормальная копия гена NOTCH3 переопределяет другую "хорошую" копию, запуская болезнь. Если ребенок получает мутировавший ген, то у него 100 % развивается синдром Цадасил. Но также известны случаи спорадического развития синдрома Цадасил без семейного анамнеза.
Синдром Цадасил вызван мутацией в гене NOTCH3 хромосомы 19q12. Ген состоит из 33 экзонов, включая 23 экзона (от 2 до 24), которые кодируют EGF-подобные домены (впервые идентифицированные в эпидермальном факторе роста) с шестью остатками цистеина. На сегодняшний день все аномалии (их более 170), ответственные за болезнь, расположены в этих экзонах. Мутации очень стереотипны, и все они приводят к добавлению или потере одного цистеина в одном из эпидермальных факторов роста. Известно, что ген NOTCH3 регулирует внутриутробную дифференцировку и созревание гладкомышечных клеток стенок мелких артерий головного мозга, мышц, кожи и внутренних органов. Он участвует в поддержании нормального функционирования сосудов и продолжительности жизни их гладкомышечных клеток [1].
Мутация в гене NOTCH3 приводит к синтезу аномального белка и неполноценному функционированию NOTCH3-рецепторов. В результате в клетках гладкой мускулатуры мелких сосудов откладываются специфические гранулярные осмиофильные отложения (их можно увидеть с помощью электронного микроскопа) и происходит дегенерация и утолщение их гладкомышечной оболочки стенки. Просвет таких сосудов не может полноценно регулироваться (повышение или снижение тонуса). Из-за этого появляются мигренозные (или мигренозоподобные) приступы и развивается хроническая ишемия (в основном в области подкорковых структур и белого вещества полушарий головного мозга) на фоне развивающейся окклюзии мелких перфорирующих сосудов. Неизвестно, как именно мутации в гене Notch3 приводят к накоплению специфических гранулярных осмиофильных отложений и дегенерации клеток, но точно установлена важная роль, которую играет данный ген в развитии патологии мелких артерий [2].
Генетически обусловленные специфические изменения стенки мелких сосудов приводят к развитию хронической гипоперфузии. Гранулярные осмиофильные включения, ответственные за утолщение средней оболочки, вызывают пролиферацию компонентов базальной мембраны с механической странгуляцией мелких артерий. Данные включения в средней оболочке приводят также к нарушению гематоэнцефалического барьера, что способствует развитию отека мозга. Кроме того, активированные ишемией астроциты в микроокружении сосудистой стенки высвобождают эндотелин-1, который вызывает вазоконстрикцию и нарушение кровотока, что является дополнительным фактором сужения мелких артерий.
Таким образом, морфологической характеристикой синдрома Цадасил является системная артериопатия с преимущественным поражением мелких церебральных артерий и артериол. Типичны концентрическое утолщение сосудистой стенки за счет субэндотелиальной фиброзной пролиферации и гиалиноза интимы, фибриноидный некроз и интрамуральный отек. При электронной микроскопии могут выявляться гранулярные осмиофильные включения вблизи гладкомышечных клеток мелких артерий. Состав гранулярных осмиофильных включений неизвестен, предполагается, что белок Notch3 является одним из их компонентов, а филаменты не входят в состав включений [4]. Указанные изменения наблюдаются не только в церебральных сосудах, но и в артериях внутренних органов (почки, печень, селезенка), а также в скелетных мышцах и коже, что позволяет использовать ультраструктурное исследование биоптатов кожи (мышцы) в качестве удобного метода прижизненной диагностики синдрома Цадасил [3].
Клинически синдром Цадасил проявляется обычно на 3–7-м десятилетии жизни повторными ишемическими инсультами (средний возраст развития инсульта составляет 49 лет) или транзиторными ишемическими атаками. Инсульты носят лакунарный характер и, что характерно, возникают обычно в отсутствие артериальной гипертонии и иных сосудистых факторов риска, имеют рецидивирующее течение, характеризуются наличием классических синдромов лакунарных инсультов и полной клинической ремиссией через несколько дней или недель (в частности, после первых инсультов).
Весьма характерными и ранними (в том числе доклиническими) признаками болезни являются изменения головного мозга, обнаруживаемые при КТ и МРТ: они включают комбинацию небольших лакунарных инфарктов в белом веществе больших полушарий и моста с диффузными изменениями белого вещества по типу лейкоареоза. Достаточно специфичным МРТ-признаком считается вовлечение полюса височной доли. Нейровизуализационное исследование при Цадасил имеет важное диагностическое значение, так как обнаруживает вышеописанные характерные изменения не только при наличии клинических симптомов заболевания, но и у всех носителей патологического гена после 35 лет, независимо от клинической картины [4].
Таким образом, резюмируя вышесказанное, для установления достоверного диагноза необходимо соответствие критериям вероятного синдрома Цадасил и выявление генетической мутации и/или артериопатии.
Основными критериями диагностики вероятного синдрома Цадасил является:
- возраст в дебюте заболевания моложе 50 лет;
- наличие хотя бы 2 из следующих клинических симптомов: инсульты, мигрень, нарушения настроения, субкортикальная деменция;
- отсутствие сосудистых факторов риска, этиологически связанных с неврологическими проявлениями;
- очевидность наследственной аутосомно-доминантной передачи; поражение белого вещества полушарий головного мозга и отсутствие кортикальных инфарктов при МРТ [5,6].
МРТ в дифференциальной диагностике синдрома-Цадасил и РС.
Были разработаны различные критерии для диагностики РС методом МРТ.На сегодняшний день используют МРТ-критерии McDonald (2017)по имени одного из авторов экспертной группы.
МРТ-критерии диссеминации процесса в пространстве – КРИТЕРИИ McDonald (2017)
Наличие >/1 очага в двух из четырех типичных локализаций:
|
1. |
>/1 очагов перивентрикулярно; |
|
2. |
>/1 очага юкстакортикально/ кортикально; |
|
3. |
>/1 очага инфратенториально; |
|
4. |
>/1 очага в спинном мозге. |
Не имеет значение симптомность очагов в стволе мозга, спинном мозге и зрительном нерве.
МРТ-критерии диссеминации процесса во времени – КРИТЕРИИ McDonald (2010, 2017)
|
1. |
Одновременное наличие накапливающих и не накапливающих контрастное вещество очагов вне зависимости от времени исследования; |
|
2. |
Появление новых очагов в режиме Т2 или накапливающих контрастное вещество очагов при повторных исследованиях вне зависимости от времени проведенного первого исследования |
|
3. |
Наличие олигоклональныхIgG в ликворе – второй тип синтеза (2017)! |
Для более полного осмысления вышеуказанных критериев далее рассмотрим типичную локализацию очагов демиелинизации:
- супратенториальные очаги: перивентрикулярныеочаги, расположенные в белом веществе, включая мозолистое тело;
- юкстакортикальныеочаги, затрагивающие U-волокна и непосредственно прилежащие к серому веществу;
- - отдельные гиперинтенсивные очаги, прилежащие к телу или височному рогу бокового желудочка, которые весьма характерны для РС и редко встречаются при других патологиях.
Говоря о характеристике очагов, нельзя не упомянуть о типах накопления контрастного вещества, которое бывает однородным (у острых, активных очагов); кольцевидным (у хронических, активных очагов); накопление по типу «полукольца» (у хронических, активных очагов) и, наконец, отсутствие накопления парамагнетика, что встречается у хронических, неактивных очагов [7,8,9].
В повседневной практике встречаются ситуации, когда при отсутствии специфической неврологической симптоматики присутствует специфическая МРТ картина, соответствующая РС, основываясь на диагностических МРТ критериях. В данном случае говорят о радиологически изолированном синдроме (РИС). Поэтому понятным становится значимость использования МРТ критериев диагностики РС при предполагаемом РИС.И, тем более, не в каждом случае наличия каких-либо очагов в головного мозге, не соответствующих МРТ критериям диагностики РС, следует думать о РИС.
В этой связи CharilA. etal., в 2006 разработали МРТ-признаки, которые могут свидетельствовать об альтернативном рассеянному склерозу диагнозе или МР-сигналы опасности, в случае нахождения которых, необходимо в первую очередь исключить нижеуказанные состояния:
Без очаговых изменений: оптикомиелит (болезнь Девика), острый поперечный миелит.
Очаги больших размеров: атипичные формы РС, первичный артериит ЦНС (сопровождающийся масс- эффектом).
Симметрично расположенные очаги: ОРЭМ, лейкоэнцефалопатии взрослых.
Нечеткие края очагов: ОРЭМ.
Отсутствие или малое количество очагов в виде «пальцев Доусона», очагов в мозолистом теле и очагов, расположенных преимущественно перивентрикулярно: ОРЭМ.
Отсутствие новых очагов при последующих наблюдениях: ОРЭМ.
Гиперинтенсивный МР-сигнал в режиме Т2-ВИ в белом веществе височных долей, островка, в области U- волокон и наружных капсул: синдром Цадасил.
Множественные двусторонние очаги с микрогеморрагиями: синдром Цадасил.
Отсутствие очагов в мозолистом теле и белом веществе мозжечка: Цадасил микроангиопатия.
Очаги, расположенные преимущественно в мозолистом теле: синдром Сусака.
Кровоизлияния: первичный артериит ЦНС, болезнь Харста.
Накопление контрастного вещества всеми очагами: ОРЭМ, первичный артерии ЦНС.
Единичные накопления контрастного вещества: первичный артериит ЦНС, саркоидоз, болезнь Бехчета.
Одиночные очаги, накапливающие контрастное вещество по типу «кольца» (чаще замкнутого): абсцессы, новообразования, первичнаялимфома ЦНС.
Инфаркты: системные аутосомные заболевания, первичный артериит ЦНС, микроангиопатия.
Преимущественное расположение очагов в области корково-подкорковой локализации: системные аутоиммунные заболевания.
Диффузное вовлечение белого вещества: болезнь Бехчета, энцефалит (например, ВИЧ-энцефалит), микроангиопатия, синдром Цадасил.
Тромбоз венозных синусов головного мозга: болезнь Бехчета.
Крупные и инфильтрирующие ствол мозга очаг: болезнь Бехчета.
Вовлечение передних отделов височных и базальных отделов лобных долей, сопровождающиеся контрастным усилением и/или масс-эффектом: энцефалит (вызванные вирусом простого герпеса).
Наличие масс-эффекта: абсцессы, новообразования.
Мультифокальные асимметричные очаги, образующиеся сначала в субкортикальных отделах белого вещества и прогрессивно увеличивающиеся в размерах: прогрессирующая мультифокальная лейкоэнцефалопатия (ПМЛ).
Обширные поражения без масс-эффекта или редко с ним: ПМЛ.
Изолированные обширные билатеральные поражения перивентрикулярного белого вещества: дефицит меди, витамина В12.
Корково-подкорковые патологические зоны, не соответствующие ни одному сосудистому бассейну: MELAS.
Преимущественное вовлечение в патологический процесс серого вещества по сравнению с белым: энцефалит.
Двустороннее поражение: ОРЭМ (на границе серого и белого вещества), Цадасил.
Лакунарные инфаркты: Цадасил, микроангиопатия.
Гиперинтенсивный МР-сигнал в режиме Т1-ВИ в таламусах: болезнь Фабри.
Множественные отдельные очаги в базальных ядрах и таламусах: синдром Сусака.
Большие и инфильтрирующие очаги в базальных ганглиях: болезнь Бехчета.
Крупные очаги, сопровождающиеся отеком: оптикомиелит (болезнь Девика), ОРЭМ, острый поперечный миелит, синдром Шегрена.
Диффузное изменение МР-сигнала от задних столбов: дефицит меди, витамина В12.
Лакунарные инфаркты в мосту: Цадасил, микроангиопатия.
Диффузное повышение пика лактата при МР-спектроскопии: MELAS.
Накопление контрастного вещества мозговыми оболочками: синдром Сусака, первичный артериит ЦНС, болезнь Бехчета, менингит, болезнь Лайма, саркоидоз.
Отсутствие поражения зрительного нерва: ПМЛ.
В заключении хотелось бы еще раз напомнить, что РС -это клинический диагноз, который ставится врачом- неврологом на основании комплексного обследования пациента. МРТ является основным методом подтверждающим клинический диагноз РС, соответственно знания и опыт специалиста, который интерпретирует полученные изображения имеют решающее значение для постановки правильного диагноза, что накладывает на него дополнительную ответственность и требует постоянного самосовершенствования в визуализации соответствующей патологии [10,11].
Клинический случай. В октябре 2020 года в неврологическое отделение поступил мужчина, 45 лет, с жалобами на затруднение при ходьбе, быструю утомляемость, самостоятельно может пройтись до 100 метров с передышками, снижения зрения с эпизодом внезапной потери зрения на 2 часа слева, охриплость голоса в течении месяца, периодические головные боли, головокружения, снижения памяти и внимания.
Из анамнеза. Считает себя больным с 6го октября 2016 года, когда внезапно начали беспокоить слабость и онемения левых конечностей, повышение АД до 180/120мм.рт.ст. Вызвал СМП, был доставлен в стационар. Проведено МРТ-исследование головного мозга: дисциркуляторная энцефалопатия с умеренной атрофией полушария ГМ. Отпущен домой на амбулаторное лечение. На следующий день в связи с углублением ухудшения состояния вызвали СМП, был доставлен в стационар в оглушенном состоянии и с левосторонним гемипарезом. Проведено КТ-исследование головного мозга, где обнаружено ОНМК по ишемическому типу в правой гемисфере. ДЭП. Госпитализирован в инсультное отделение, и диагноз: ОНМК по ишемическому типу в бассейне правой среднемозговой артерии. Гемипарез слева. АГ 3ст, риск 4. СНФК 2ст. Получил антикоагулянтную антигипертензивную терапию. Выписался с левосторонним гемипарезом, силой мышц в левых конечностях – 3,5 баллов. После в течении около 4х месяцев движение и сила левых конечностей
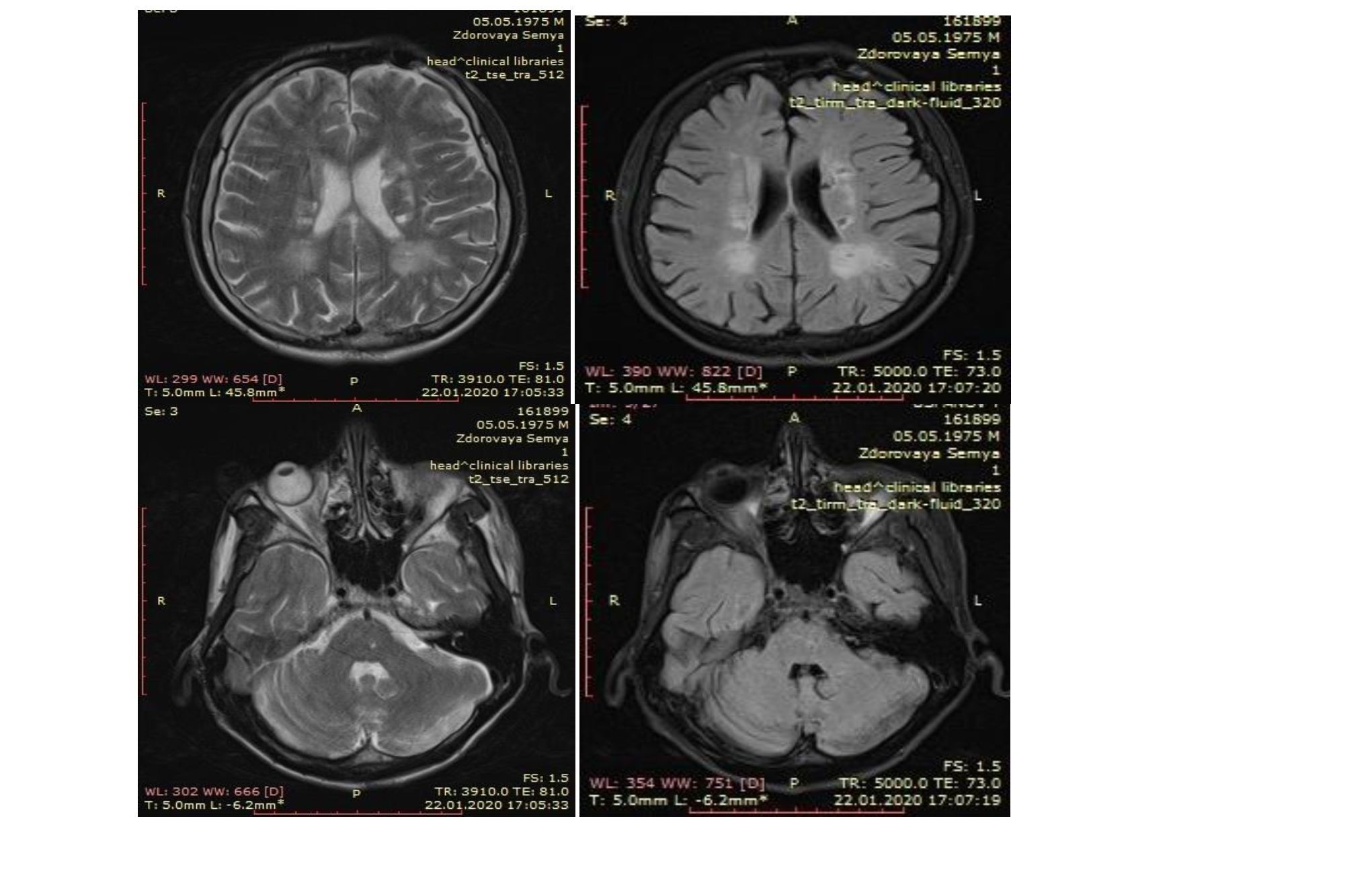
полностью восстановились. Периодические головные боли повторялись. В 2019 году 6 июня на фоне высокого АД 190/110мм.рт.ст. появились онемения левых конечностей, нарушение речи. Вызвали СМП, поступил в инсультное отделение в оглушенном состоянии. Проведено МРТ головного мозга: ОНМК по ишемическому типу в правой гемисфере. Был выставлен диагноз: ОНМК по ишемическому типу в бассейне правой среднемозговой артерии. Гемипарез слева. АГ 3ст, риск 4. СНФК 2ст. В стационаре получил антикоагулянтную, антиагрегентную и антигипертензивную терапию. Выписался с дизартрией, асимметрией лица и левосторонним гемипарезом. Полный регресс неврологической симптоматики заняло около 3-4 месяцев. В ноябре 2019 года во время планового МРТ-обследования почувствовал ухудшение состояния в виде онемения конечностей, языка, нарушения речи, повышения АД до 160/100мм.рт.ст. Проведено МРТ головного мозга, где обнаружено МР- признаки ишемического микроинсульта левого полушария головного мозга, в обеих половинах зрительного бугра в стадии образования кист, дисциркуляторной энцефалопатии. Сосудистой энцефалопатии с наличием ишемического участка в проекции таламуса. Госпитализирован в ЦРБ, где получил антиагрегантную и антигипертензивную терапию. Был выставлен диагноз: ОНМК по ишемическому типу в бассейне правой среднемозговой артерии. Гемипарез слева. АГ 3ст, риск 4. СНФК 2ст. Выписался с дизартрией, асимметрией лица и левосторонним гемипарезом, гипомнезией. Неврологическая симптоматика постепенно регрессировала.
С начала 2020 года начал отмечать изменение походки и быструю утомляемость, симптоматика постепенно нарастала, присоединись снижения памяти и внимания. В апреле 2020 года обратился за консультацией к невропатологу, где заподозрили рассеянный склероз. Проведено МРТ головного мозга: МР- картина наиболее характерна для демиеленизирующего заболевания (РС) с множеством очагов демиелинизации субкортикально, перивентрикулярно, в базальных ядрах БШ ГМ, а так же в структуре моста мозга, с «черными дырами», наиболее выраженными в базальных ядрах размерами до 0,91х1,05см, с признаками накопления парамагнетика по краю «черных дыр» в участках демиелинизации в базальных ядрах обоих полушарий головного мозга, а так же участком в структуре моста мозга размерами до 0,29х0,32см. Церебральная атрофия легкой степени. Внутричерепная гипертензия. Субатрофия коры обеих гемисфер мозжечка. Кистовидное расширение большой цистерны мозга размерами до 1,22х2,32х2,32см – mega cisterna magna.
91
В неврологическом статусе: сознание ясное. Зрачки Д=S. Легкая асимметрия н/г складок. Язык по средней линии. В позе Ромберга не устойчив. Пальце-носовая проба –с интенцией 2-х сторон. Сила мышц верхних конечностях 4 балла, в нижних конечностях 3,5-4,0 балла. Гипертонус задних икроножных мышц справа, Шаркающая походка (Походка с мелкими шагами) Сухожильные рефлексы Д<S, гиперрефлексия слева, клонусы при коленном СХР слева.
Результаты лабораторных и инструментальных исследований.
В общеклинических анализах, анализе цереброспинальной жидкости (ЦСЖ) каких-либо отклонений от нормы не выявлено. Анализ ЦСЖ на микобактерии туберкулеза, атипичные клетки, антитела к цитомегаловирусу отрицательный. Ревматологические пробы отрицательные. Анализ уровня антител IgG, IgM к фосфолипидам — норма, антинуклеарные антитела CLIA в крови отсутствуют. Реакция Вассермана отрицательная. Антитела к ВИЧ не обнаружены. Уровень лактата крови — 0,8 ммоль/л (норма 0,5–2,2 ммоль/л). ЭКГ-ритм синусовый, регулярный. Были взяты анализы на олигоклональные антитела и антитела к аквопорину-4, которые были отрицательными. После проведенного комплексного обследования пациента диагноз рассеянный склероз был снят. Наконец, был получен геннетичекий анализ, где были обнаружены точечные мутации в гене NOTCH3 во 2-3 экзонах на 19-й хромосоме, что подтверждает данный диагноз. Учитывая вышеизложенные данные, выставлен диагноз: церебральная аутосомно-доминантная артериопатия с субкортикальными инфарктами и лейкоэнцефалопатией, синдром CADASIL.
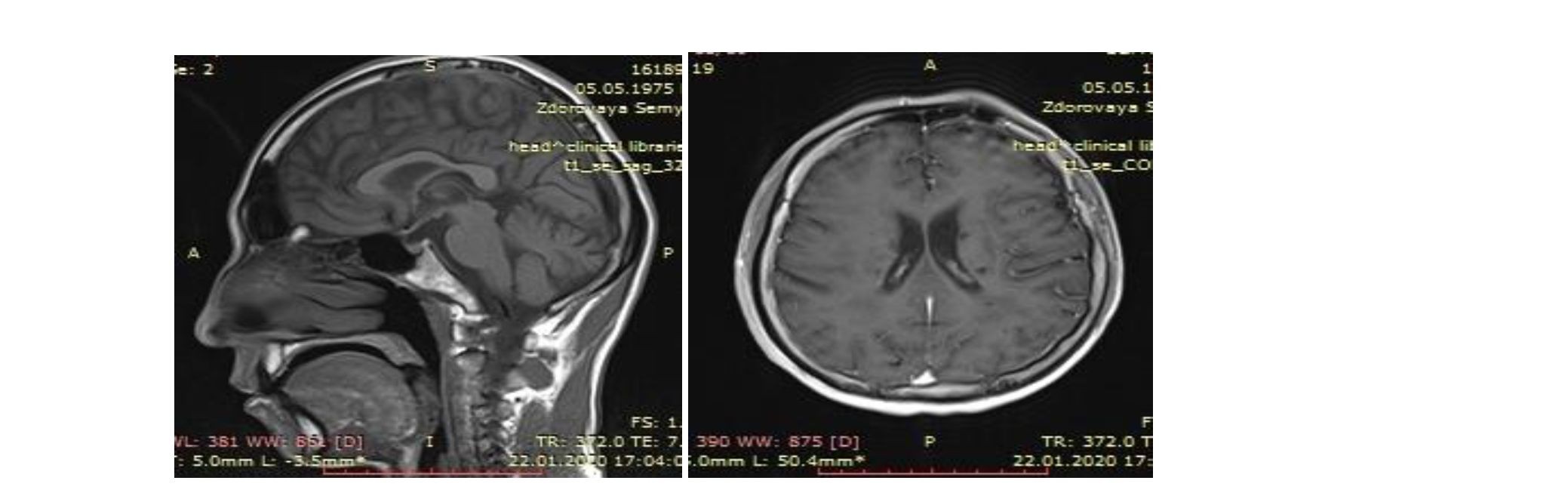
Выводы: Таким образом, для установления достоверного диагноза синдрома Цадасил необходимо соответствие критериям вероятной Цадасил и выявление генетической мутации и/или артериопатии. В настоящее время предполагается, что синдром Цадасил в большинстве случаев не диагностируется. В данном клиническом случае, из выстроенного дифференциального ряда наши врачи исключили заболевания демиелинизирующего характера и нейроинфекцию. Окончательная диагностика выявила МР-признаки, характерные именно для ЦАДАСИЛ, а именно: относительно молодой возраст пациента, локализация изменений в полюсах височной, передних отделах лобных долей, поражение внутренней капсулы, перивентрикулярного белого вещества и лейкоэнцефалопатии, а в последующем, диагноз был подтвержден генетическим анализом.
Список литературы
- Кротенкова М.В., Брюхов В.В., Морозова С.Н., Кротенкова И.А., Магнитно резонансная томография в диагностике и дифференциальной диагностике рассеянного склероза. ГЭОТАР- Медиа, 2019 г.
- Barkhof F., Filippi M., Miller D.H. et al. Comparison of MR imaging criteria at first presentation to predict conversion to clinically definite multiple sclerosis // Brain. — 1997. — 120. — 20592069.
- Lublin F.D. Predicting the Course of Multiple Sclerosis: Implications for Treatment // Medscape Education Neurology & Neurosurgery. — 2011.
- Fazekas F., Barkhof F., Filippi M. et al. The contribution of magnetic resonance imaging to the diagnosis of multiple sclerosis // Neurology. — 1999. — 53. — 448456.
- Kornek B., Schmitl B., Vass K. Evaluation of the 2010 McDonald multiple sclerosis criteria in children with a clinically isolated syndrome // Multiple Sclerosis Journal. — 2012. — 18 (9). — 112119.
- McDonald I.P., Compston A., Edan G. et al. Recommended Diagnostic Criteria for Multiple Sclerosis: Guidelines from the International Panel on the Diagnosis of Multiple Sclerosis // Ann. Neurol. — 2001. — 50. —121127. 92
- Paty D.W., Oger J.J., Kastrukoff L.F. et al. MRI in the diagnosis of MS: a prospective study with comparison of clinical evaluation, evoked potentials, oligoclonal banding and CT // Neurology. — 1988. — 38. — 180185.
- Polman C.H., Reingold S.C., Banwell B. et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria // Annals of Neurology. — 2011. — 69 (2). — 292302.
- Polman C.H., Reingold S.C., Edan G. et al. Diagnostic Criteria for Multiple Sclerosis: 2005 Revisions to the «McDonald Criteria» // Annals of Neurology; Published Online: November 10, 2005.
- Poser C.M., Paty D.W., Scheinberg L. et al. New diagnostic criteria for multiple sclerosis: guidelines for research protocols // Ann. Neurol. — 1983. — 13. — 227231.
- Thompson A.J., Montalban X., Barkhof F. et al. Diagnostic criteria for primary progressive multiple sclerosis: a position paper // Ann. Neurol. — 2000. — 47. — 831835.
References
- Krotenkova M. V., Bryukhov V. V., Morozova S. N., Krotenkova I. A., Magnitnoresonant tomography in the diagnosis and differential diagnosis of multiple sclerosis. GEOTAR-media, 2019
- Barkhof F., Filippi M., Miller D.H. et al. Comparison of MR imaging criteria at first presentation to predict conversion to clinically definite multiple sclerosis // Brain. — 1997. — 120. — 20592069.
- Lublin F.D. Predicting the Course of Multiple Sclerosis: Implications for Treatment // Medscape Education Neurology & Neurosurgery. — 2011.
- Fazekas F., Barkhof F., Filippi M. et al. The contribution of magnetic resonance imaging to the diagnosis of multiple sclerosis // Neurology. — 1999. — 53. — 448456.
- Kornek B., Schmitl B., Vass K. Evaluation of the 2010 McDonald multiple sclerosis criteria in children with a clinically isolated syndrome // Multiple Sclerosis Journal. — 2012. — 18 (9). — 112119.
- McDonald I.P., Compston A., Edan G. et al. Recommended Diagnostic Criteria for Multiple Sclerosis: Guidelines from the International Panel on the Diagnosis of Multiple Sclerosis // Ann. Neurol. — 2001. — 50. —121127.
- Paty D.W., Oger J.J., Kastrukoff L.F. et al. MRI in the diagnosis of MS: a prospective study with comparison of clinical evaluation, evoked potentials, oligoclonal banding and CT // Neurology. — 1988. — 38. — 180185.
- Polman C.H., Reingold S.C., Banwell B. et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria // Annals of Neurology. — 2011. — 69 (2). — 292302.
- Polman C.H., Reingold S.C., Edan G. et al. Diagnostic Criteria for Multiple Sclerosis: 2005 Revisions to the «McDonald Criteria» // Annals of Neurology; Published Online: November 10, 2005.
- Poser C.M., Paty D.W., Scheinberg L. et al. New diagnostic criteria for multiple sclerosis: guidelines for research protocols // Ann. Neurol. — 1983. — 13. — 227231.
- Thompson A.J., Montalban X., Barkhof F. et al. Diagnostic criteria for primary progressive multiple sclerosis: a position paper // Ann. Neurol. — 2000. — 47. — 831835.