Резюме
Среди существующих в хроматографии типов детекторов масс-спектрометрические выигрышно выделяются по своей селективности, и иногда возникает ощущение, что современное оборудование само по себе способно избавить аналитика от множества проблем при разработке методики. Однако примерно в каждом пятом случае можно наблюдать недостаточную специфичность методики. При этом руководства по валидации биоаналитических методик не дают алгоритма определения специфичности. Целесообразно на этапе валидации методики установить критерии пригодности хроматографической системы и с помощью методов математической статистики рассчитать допустимые интервалы значений этих параметров. При дальнейшей рутинной работе эти нормы помогут своевременно выявить случаи несоответствий, выяснить причины отклонений и принять меры по доработке методики.
Ключевые слова: ВЭЖХ-МС/МС, селективность, специфичность, валидация.
Метод ВЭЖХ-МС/МС прочно вошёл в рутинную практику проведения биоаналитических исследований, существенно облегчив и упростив не только процесс разработки методики, но и интерпретацию результатов. Процедура валидации обязательно завершает этап разработки методики, позволяет подтвердить её надежность и применимость для последующих рутинных исследований.
Среди основных характеристик, определяемых при валидации биоаналитических методик, особое место занимают селективность и специфичность, которые устанавливают едва ли не в первую очередь, т.к. без их подтверждения невозможна дальнейшая валидация, а неселективная и неспецифичная методика требует доработки.
Безусловно, среди существующих в хроматографии типов детекторов масс-спектрометрические выигрышно выделяются по своей селективности, и иногда возникает ощущение, что современное оборудование само по себе способно избавить аналитика от множества проблем при разработке методики.
Современные руководства по валидации биоаналитических методик [1-3] определяют селективность как способность дифференцировать исследуемый аналит от эндогенных компонентов матрицы и других компонентов образца. На практике с этой целью проводят сравнение хроматограмм интактной матрицы с хроматограммами модельных образцов, которые содержат как компоненты матрицы, так и аналит. В случае отсутствия сигнала аналита на хроматограмме интактного образца методика признаётся селективной.
Однако при последующем рутинном анализе образцов от добровольцев или пациентов могут возникать самые разные ситуации. Например, на хроматограмме неожиданно может появиться какой-либо посторонний пик, либо на пике аналита появится "наездник", либо время удерживания и симметрия пика будут отличаться от того, что было получено на этапе валидации. В связи с этим, необходимо дополнительно определять такую характеристику методики, как специфичность, т.е. способность однозначно измерять аналит в присутствии других соединений (эндогенных или экзогенных) в матрице. Очевидно, что это определение дано очень широко, и нельзя сразу понять, о каких конкретно соединениях идёт речь. Руководство FDA 2018 г и Правила ЕАЭС 2015 г уточняют [2,3], что в первую очередь речь идёт о метаболитах изучаемого аналита и других веществах, присутствие которых возможно в образце (например, другие лекарственные средства). Однако проблема состоит в том, что ни одно руководство не даёт алгоритма определения специфичности, а аналитик не может знать полный состав образца, чтобы на этом основании делать какие-либо прогнозы. Таким образом, необходимо иметь некую стратегию и методологию разработки и валидации биоаналитических методик, которые позволят достигнуть максимальной селективности и специфичности.
По нашим наблюдениям, проблема недостаточной специфичности методики возникает практически в каждом пятом случае несмотря на использование МС-детектора.
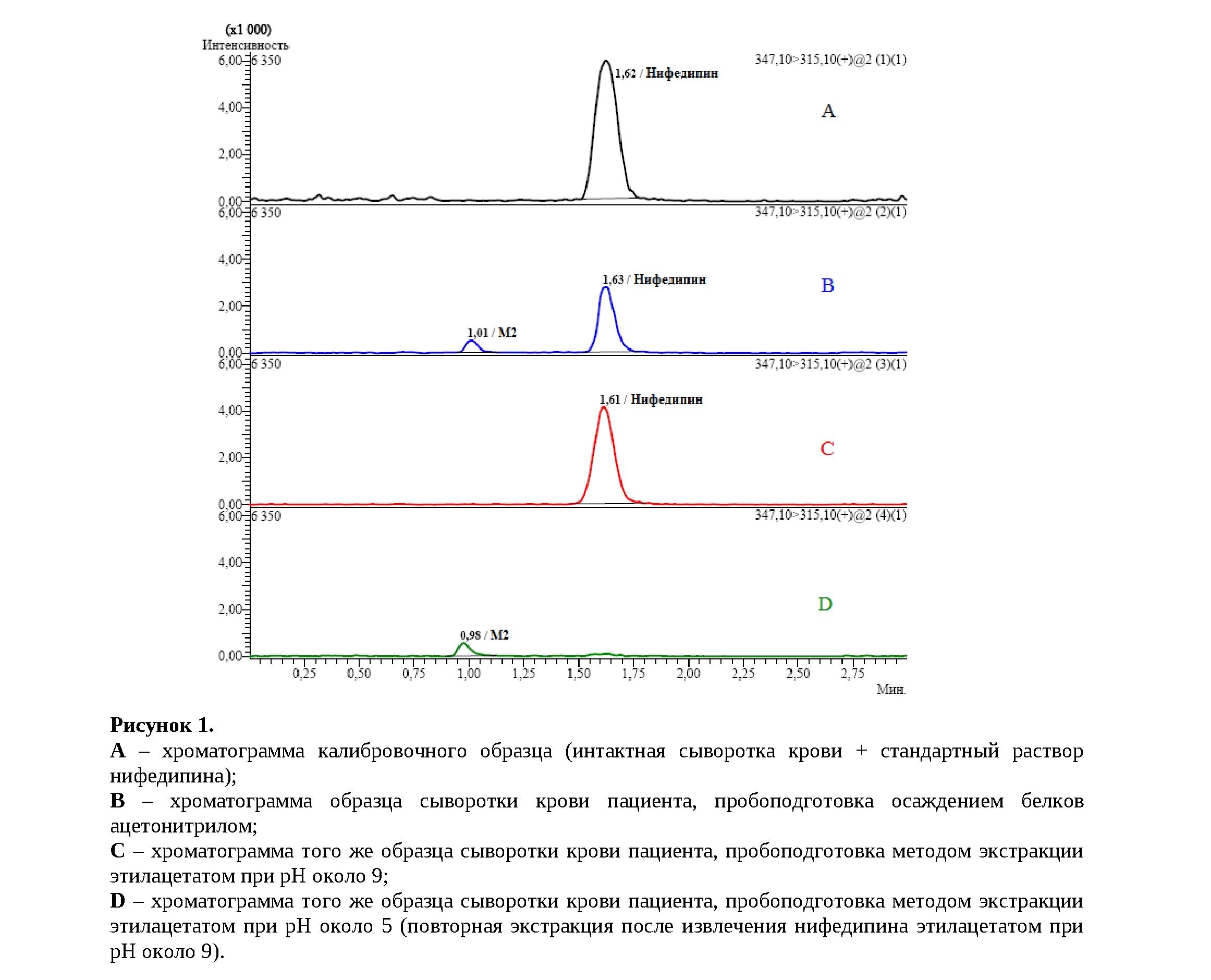
Например, при рутинном анализе проб сыворотки пациентов, принимавших нифедипин, с помощью разработанной и валидированной нами ранее методики[4] было сделано любопытное наблюдение. В одних случаях хроматограмма для нифедипина имела такой же вид, как у калибровочных образцов (Рисунок 1A) и образцов контроля качества (QC ‒ quality control), но в других случаях на хроматограмме наблюдалось два пика (Рисунок 1B). Один из них соответствовал по времени удерживания нифедипину, а второй пик относился к неизвестному веществу, которое удерживалось слабее нифедипина, при этом пики были хорошо разделены хроматографически.
Для объяснения наблюдаемого явления мы выдвинули две гипотезы. Во-первых, наличие 2-х пиков могло быть связано с какими-либо проблемами хроматографического разделения, например, усталость/загрязнение сорбента колонки и/или предколонки, и в этом случае оба пика могут принадлежать нифедипину, элюирующемуся в виде двух зон. Во-вторых, возможна интерференция с каким-то другим веществом в составе образца. При этом следует учитывать, что масс-спектрометрический детектор работал в режиме MRM+ (multiple reaction monitoring, мониторинг множественных реакций) с регистрацией перехода 347,1→315,1, то есть использовался высокоселек-тивный режим работы, и риск интерференции с каким-либо посторонним соединением нам казался крайне низким, т.к. для этого неизвестное вещество должно иметь не только такую же молекулярную массу (М.м.), что и аналит, но и максимально близкую структуру, чтобы иметь аналогичный путь фрагментации.
4
Первая гипотеза была отвергнута по следующим причинам:
- во-первых, 2 пика присутствовали лишь на части хроматограмм проб пациентов, тогда как для других образцов, проанализированных в рамках того же аналитического цикла, "второй- неизвестный" пик отсутствовал, причём его никогда не было на хроматограммах калибровочных и QC образцов;
- во-вторых, оставались постоянными время удерживания и асимметрия основного пика, хотя их изменения следует ожидать в случаях ухудшения условий хромато-графического разделения.
В связи с выше сказанным, вторая гипотеза о наличии в образцах неустановленного компонента со сходной структурой и соответствующей нифедипину молекулярной массой была принята за основную.
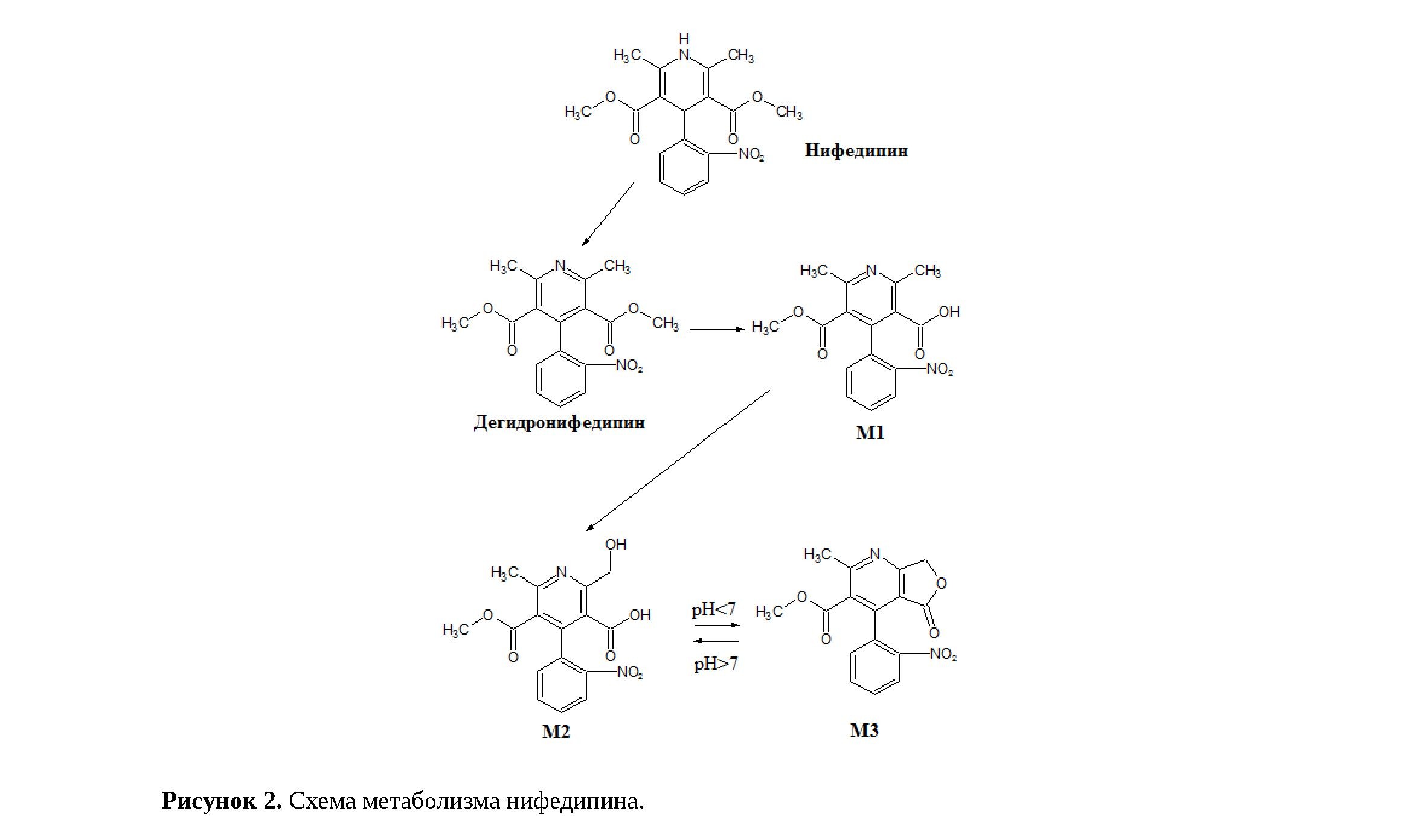
При внимательном изучении информации [5] о биотрансформации нифедипина было обнаружено, что у него существует метаболит М2 (Рисунок 2), М.м. которого (346,29) практически точно соответствует М.м. нифедипина (346,33). MRM-переход, использованный для детектирования нифедипина (347,1→315,1) обусловлен отщеплением СН3ОН от [M+H]+. Структура метаболита также даёт возможность для такой фрагментации.
Поскольку стандартного образца предполагаемого метаболита для прямого доказательства нашей гипотезы у нас не было, закупка была бы длительной по времени, а ответ нужно было получить срочно, то для подтверждения/опровержения нашей второй гипотезы были проведены дополнительные мероприятия.
Нифедипин не обладает выраженными кислотными или основными свойствами, причём даже нет достоверной непротиворечивой информации о его рКа. Метаболит М2, напротив, содержит карбоксильную группу, придающую кислотные свойства, а также слабоосновный пиридиновый атом азота. Таким образом, химические свойства нифедипина и М2 радикально отличаются, и эти вещества можно разделить на этапе пробоподготовки образца сыворотки методом жидкость-жидкостной экстракции. Действительно, при основных значениях рН в органическую фазу извлекался только сам нифедипин (Рисунок 1C), метаболит М2 в ионизированной форме оставался в водной фазе, откуда экстрагировался после подкисления до рН около 5 (Рисунок 1D). Эти эксперименты в сочетании с литературными данными являются весомыми доказательствами того, что два пика на хроматограмме нифедипина соответствуют разным веществам, а "неизвестный" пик с большой вероятностью относится к метаболиту М2.
 5
5
Описанный пример ярко демонстрирует, что, несмотря на высокую селективность детектирования в режиме MRM, всегда существует риск, что в образце окажутся неизвестные соединения, имеющие близкую молекулярную массу и/или строение, что приведет к перекрестному детектированию. В представленном примере пики нифедипина и М2 были полностью разделены, поэтому результаты количественного определения достоверны. В других ситуациях недостаточное разрешением пиков, появление пиков-наездников и ситуация "пик в пике" приводят к неадекватному интегрированию, вследствие чего может быть получен завышенный результат по концентрации аналита. Для предупреждения указанных случаев целесообразно на этапе валидации методики установить критерии пригодности хроматографической системы (в первую очередь, время удерживания аналита и коэффициент асимметрии пика) и с помощью методов математической статистики рассчитать допустимые интервалы значений этих параметров. При дальнейшей рутинной работе эти нормы помогут своевременно выявить случаи несоответствий, выяснить причины отклонений и принять меры по доработке методики.
В приведённом примере был показан один из алгоритмов действий, которым можно воспользоваться для подтверждения специфичности методики. Таким образом вдумчивый и всесторонний анализ способен объяснить практически любые нестандартные ситуации при использовании метода ВЭЖХ-МС/МС в биоаналитических исследованиях.
Список литературы
- Руководство по экспертизе лекарственных средств / Под. ред. проф. А.Н. Миронова. Том I. – М.: Гриф и К, 2013. 328 с.
- Правила проведения исследований биоэквивалентности лекарственных препаратов в рамках Евразийского экономического союза (утверждены распоряжением №178 Коллегии Евразийской экономической комиссии от 25.12.2015 г.
- Guidance for Industry: Bioanalytical method validation. U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evolution and Research (CDER). U.S. Government Printing Office: Washington, DC, 2018.
- Родина ТА, Мельников ЕС, Белков СА, Данько АА, Соколов АВ, Прокофьев АБ. Терапевтический лекарственный мониторинг нифедипина методом ВЭЖХ-МС/МС при лечении артериальной гипертензии // Биомедицина. ‒ 2017. ‒ №4. ‒ С. 53-69.
- Raemsch K. D., Sommer J. Pharmacokinetics and metabolism of nifedipine //Hypertension. – 1983. – Т. 5. – №. 4 Pt 2. – Pp. II18.