В данной статье приведены результаты молекулярно-генетического исследования пациентов с диагностированной эпилепсией. Целью данной работы было определение ассоциации мутаций генов натриевого и калиевого каналов с развитием эпилепсии. Для проведения молекулярно-генетического анализа были выбраны 44 пациента с разными формами каналопатической эпилепсии, которые находились на лечении в SVS клинике имени В.М. Савинова. Генотипирование проводили методами сайт-специфической ПЦР-ПДРФ по гену натриевого (SCN1A) и калиевого (KCNT1) каналов. Для установления наследуемых мутаций генов SCN1A и KCNT1 проводили молекулярно-генетический анализ родственников пациентов ближайшей степени родства. В результате установлены de novo мутации в гене натриевого канала (SCN1A p.Ala1783Thr) у одного пациента с синдромом Драве. Было обнаружено 3 случая de novo мутации гена калиевого канала (KCNT1 p.Ala934Thr). Из них 2 пациента, страдали височной эпилепсией, которые имели парциальные и генерализованные приступы психомоторного автоматизма. И один пациент болел резидуальной энцефалопатией с первично генерализованными судорогами.
Введение: Эпилепсия является одним из наиболее распространенных и гетерогенных неврологических заболеваний. Основными клиническими признаками заболевания являются повторяющиеся симптоматические или идиопатические эпилептические припадки как судорожного, так и бессудорожного характера, которые развиваются на фоне утраченного или сохраненного сознания.
Согласно Кодекса Республики Казахстан от 18 сентября 2009 года «О здоровье народа и системе здравоохранения» (ст. 7, пп. 89) эпилепсия относится к социально-значимым заболеваниям. Это заболевание является одним из наиболее распространенных серьезных неврологических расстройств, которым страдают около 1% людей во всем мире (50 млн.) [1]. В Казахстане эпилепсией болеют более 45 тыс. человек, из них 40% составляют дети, подростки и молодые люди,
Таблица 1 - Клиническая характеристика пациентов
|
Количество пациентов |
44 |
|
Средний возраст |
34±11,15 |
|
(год рождения) |
(1954-2011гг) |
|
Мужской пол |
27 |
|
Женский пол |
17 |
|
Данные ЭЭГ |
|
|
Патологический вариант ЭЭГ |
38 |
|
Плоский вариант ЭЭГ |
6 |
38% больных становятся инвалидами, качество жизни снижается у них в среднем на 85% [2]. Основные перспективы в снижении таких высоких показателей заболеваемости и смертности связаны с созданием и совершенствованием методов диагностики, обладающих научно-обоснованной эффективностью. В связи с этим целью данной работы является определение ассоциации мутаций натриевого и калиевого каналов с развитием эпилепсии в Казахстане. Это позволит не только определить характер распространения наследственных эпилептических синдромов в нашей стране, но и обогатить общемировые знания о молекулярной генетике наследуемых форм эпилепсии.
В многофакторном патогенезе эпилепсии принимают участие как наследственные, так и приобретенные, опосредованные влиянием среды, факторы. Молекулярные механизмы, лежащие в основе различных эпилептических приступов, интенсивно изучаются уже более двух десятилетий. Генетическая компонента играет большую роль в этиологии идиопатических форм эпилепсии. Приблизительно 20-30% случаев эпилепсии обусловлены приобретенными состояниями, такими как инсульт, опухоль или травма головы. Однако, данные последних лет свидетельствуют, что остальные 70-80% случаев имеют генетическую природу [3].
Большинство наследственных форм эпилепсий с установленными генами обусловлены повреждениями ионных каналов, обеспечивающих поляризацию мембраны нейронов. Такие формы эпилепсии относят к группе каналопатий. К ним относят, прежде всего, гены натриевого, калиевого, кальциевого и хлорного каналов (SCN1A, SCN2A, CACNA1A, KCNJ10, KCNQ2) [4-9].
Мутации и делеции в генах натриевого канала - SCN1A, описаны для 70% детей с синдромом Драве, большая часть мутаций имеет спонтанный характер [4; 6]. Мутации SCN1A могут быть причиной развития тяжелой миоклонической эпилепсии младенчества (SMEI), относящейся к симптоматическим формам [10]. К аутосомно-доминантной ночной лобной эпилепсии (ADNFLE) и злокачественно мигрирующим парциальным приступам младенчества (MMPSI) приводят доминантные мутации в гене натрийкалиевого канала KCNT1, интенсивно экспрессирующемся в головном мозге [7]. Нарушения в этом гене способствуют повышению проницаемости мембраны, что приводит к нерегулируемому возбуждению нейронов в головном мозге. Материалы и методы:
Формирование когорты для исследования
Для молекулярно-генетического анализа на базе SVS клиники имени В.М. Савинова были собраны образцы периферической крови, полученные от пациентов (44 чел.), которым был поставлен диагноз эпилепсия в соответствии с критериями Комиссии по классификации и терминологии ILAE (1989). Перед забором крови у всех пациентов получали добровольное информированное согласие для исследования и проводили подробное анкетирование. Исследование было одобрено Этическим Комитетом Казахстанско-Российского Медицинского Университета. Выделение геномной ДНК
ДНК выделяли из замороженных (-20°С) образцов периферической крови, содержащих в качестве антикоагуляционного агента ЭДТА. Выделение проводилось с использованием набора для выделения ДНК GeneJet Genomic DNA Purification Kit (Thermo Fisher Scientific, USA) по протоколу, рекомендованному фирмой производителем. Количественную и качественную оценку препаратов ДНК проводили с помощью спектрофотометрического и электрофоретического анализа. После выделения образцы ДНК хранили при -20 °С.
ПЦР-амплификация генов SCN1A и KCNT1
ПЦР проводили со специфическими праймерами, дизайн которых был подобран с помощью онлайн программы - PrimerQuest Tool (для гена SCN1A - F- CCCGACTGTGACCCTAATAAAG, R-GTTTGGTTGTGGCAGATTGAG, для гена KCNT1-F-CACCCTGAGACCTCCTACAA, R- CCCTTTCTCCCACTCTTTCTG). Реакционная смесь для проведения ПЦР содержала: 25 ng геномной ДНК, 0,5 pmol каждого праймера, 10 p.L Mmix и 7^L H2O. ПЦР амплификацию проводили при условии, для гена SCN1A: инициирующий этап - 94°С, 4 мин., затем следует блок из 35 циклов: 94°С, 40 сек., 55°С,30 сек., 72°С, 40 сек., и этап финальной элонгации - 72 °С, 8 мин. для гена KCNT1: инициирующий этап - 95°С, 3 мин., затем следует блок из 35 циклов: 94°С, 30 сек., 58°С,30 сек., 72°С, 30 сек., и этап финальной элонгации - 72 °С, 10 мин. ПЦР проводили в 0.2 ml микропробирках на термоциклере «Eppendorf™ Mastercycler™ Nexus Thermal Cycler» с набором программ, определяющих температурный режим ПЦР. Для оценки количества и специфичности полученного в результате проведенного ПЦР, амплифицированные продукты проверяли электрофоретическим методам в 1,5 агарозном геле. Соответствие молекулярных масс амплификатов каждого гена оценивали с помощью маркера DNA Ladder GeneRuler 100 bp (ThermoFisher Scientific, USA). В качестве минус-контроля ПЦР использовали пробу, не содержащую геномную ДНК.
ПДРФ - анализ генов SCN1A и KCNT1
Полученные ПЦР продукты, подвергались гидролизу эндонуклеазой рестрикции Acc II (ThermoFisher Scientific, USA) при температуре инкубации 37°С, в течение 5 часов. Затем продукты рестрикции прогоняли в 8% полиакриламидном геле (ПААГ) и окрашивали гель бромистым этидием, полученные результаты анализировали с помощью трансиллюминатора. Оценку полученных фрагментов проводили с помощью маркера DNA Ladder GeneRuler 100 bp (Thermo Fisher Scientific, USA). При анализе продуктов рестрикции гомозиготный генотип нормального аллеля гена SCN1A представлял два фрагмента ДНК размером 188 bp и 133bp. Гомозиготный генотип мутантного аллеля представлял один фрагмент размером 321 bp. Соответственно, гетерозиготный генотип устанавливали по наличию 3-х фрагментов размером 321 bp, 188 bp и 133bp. При ПДРФ-анализе гена KCNT1 гомозиготный генотип нормальной аллели представлен двумя фрагментами размером 128 и 105 bp, гомозиготный вариант мутантного аллеля - одним фрагментом размером 233 bp, и, соответственно, гетерозиготный вариант - тремя фрагментами размером 233 bp, 128 bp и 105 bp.
Результаты и их обсуждение.
Демографические и клинические данные согласившихся на участие в исследовании 44 пациентов обобщены в таблице 1. Клиническое обследование 44 пациентов с различными формами эпилепсии, показало, что 9 человек болели височной эпилепсией, 9 человек - эпилепсией с тоникоклоническими приступами, 3 человека - ювенильной и детской абсансной эпилепсией, 5 человек - первично генерализованной эпилепсией, 4 человека - вторично генерализованной эпилепсией, 5 имели резидуальную энцефалопатию, 3 человека - ювенильную миоклоническую эпилепсию, 2 человека - ADNFLE, 4 человека идиопатическую эпилепсию.
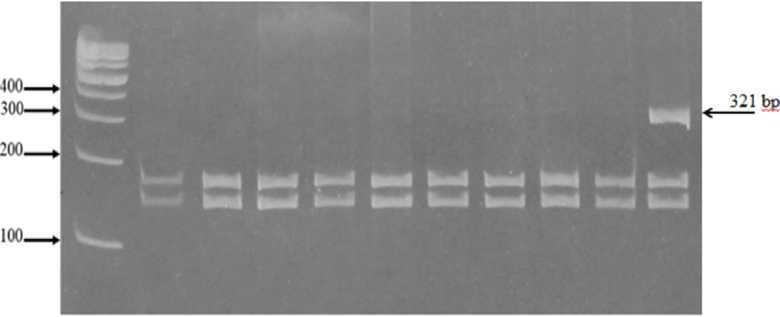
Для определения генетического статуса пациентов с диагнозом эпилепсия были исследованы гены SCN1A (26 экзон) и KCNT1 (24 экзон). Был проведен рестрикционный анализ для кодонов Ala1783Thr и p.Ala934Thr. С помощью
RFLP анализа была обнаружена мутация гена SCN1A - p.Ala1783Thr (c.5347G> A) у одного пациента (2,5 года) с синдромом Драве (рисунок 1).
|
Генерализованные приступы |
31 |
|
Парциальные приступы |
13 |
М 1 2345 б 78 9 10
M - маркер молекулярного веса 100-bp
1-9 нормальные образцы.
- - образец с гетерозиготной мутацией
Рисунок 1 - ПЦР-ПДРФ анализ мутации гена SCN1A - p.Ala1783Thr (c.5347G>A)
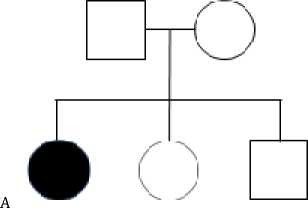
Обследования родителей и 3-месячной сестры не выявили мутации в этом кодоне (рисунок 2), что указывало на de novo появление этой мутации. Впервые судороги у пациента возникли в 3 месяца и повторялись 2 раза в месяц с разной семиотикой, фебрильные судороги не возникали.




Рисунок 2 - Семейное древо пациента с синдромом Драве
De novo мутация гена SCN1A (p.Ala1783Thr), которая привела к нарушению функции натриевого канала, является свидетельством синдрома Драве. Синдром Драве - криптогенный эпилептический синдром, который имеет черты, как фокальных, так и генерализованных приступов и при котором судороги обычно не поддаются лечению и связаны с умственной недееспособностью. Так лечение пациента Вальпроатом привело только к незначительному улучшению, в связи, с чем его заменили на Топирамат, при этом приступы стали возникать реже. Затем пациенту был назначен препарат Оксарбазепин, при котором замечалось ухудшение состояния пациента. На основании этого пациенту снова назначили Топирамат в комбинации с
Вальпроатом и дексаметазоном, но, несмотря на это судороги возникали ежедневно с миоклонией глаз и плеч [11-13].
Было обнаружено 3 случая de novo мутации гена KCNT1 кодона p.Ala934Thr. Из них 2 пациента, страдали от височной эпилепсии (1972 г. и 1988 г.), которые имели парциальные и генерализованные приступы психомоторного автоматизма. Частота приступов была 1-2 раза в месяц. У одного пациента с диагнозом резидуальная энцефалопатия (1987 г.р.) также была идентифицирована мутация в кодоне 934 (рисунок 3), этот пациент имел первично генерализованные судороги, частота приступов была 1 раза в 1-2 месяца.
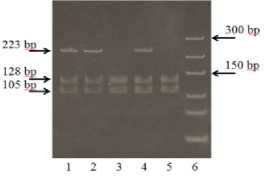
- - маркер молекулярного веса 100-bp
- 2 и 4 - образцы с гетерозиготной мутацией
- и 5 нормальные образцы
Рисунок 3 - ПЦР-ПДРФ анализ мутации гена KCNT1 934 кодон - c.2800G>A (p.Ala934Thr)
Молекулярно-генетический анализ близких родственников пациентов с мутацией в гене KCNT1 (p.Ala934Thr)
подтвердил de novo происхождение этой мутации во всех 3 семьях (рисунок 4).

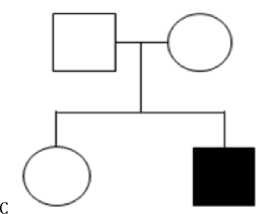
A - пациент с височной эпилепсией (год рождения 1988), B - пациент с височной эпилепсией (год рождения 1972),
C - пациент с резидуальной энцефалопатией (год рождения 1987)
Рисунок 4 - Семейное древо пациенто с de novo мутацией в гене KCNT1
Как известно из литературных источников [14,15], мутация 934 кодонов гена калиевого канала KCNT1 должна быть специфичной для злокачественно мигрирующих парциальных приступов младенчества (MMPSI). Но мы идентифицировали de novo мутацию KCNT1 (p.Ala934Thr) у пациентов, страдающих височной эпилепсией (TLE), и эта мутация не была обнаружена у пациентов ADNFLE.
Таким образом, мы можем заключить, что мутации гена калиевого канала KCNT1 могут вызывать не только аутосомно-доминантную ночной эпителиальной лобной доли (ADNFLE), но и другие формы эпилепсии.
В будущем мы планируем увеличить число пациентов и провести секвенирование генов SCN1A и KCNT1.
СПИСОК ЛИТЕРАТУРЫ
- WHO URL: http://www.who.int/mediacentre/factsheets/fs999/ru/-uHTepHeT источники
- Неврология URL: http://neurology-help.kz/eilepsija_set.html
- Hildebrand M.S., Dahl H.H., Damiano J.A., Smith R.J., Scheffer I.E., Berkovic S.F. Recent advances in the molecular genetics of epilepsy // J Med Genet. - 2013. - Vol.50. - P. 271-279.
- Scheffer I.E., Iona X., Zuberi S.M., Birch R., McMahon J.M., Bruce C.M., Berkovic S.F., Mulley J.C. De novo SCN1A mutations in Dravet syndrome and related epileptic encephalopathies are largely of paternal origin // J Med Genet. - 2010. - Vol.47. - P.137-141.
- Poryo M., Clasen O., Oehl-Jaschkowitz B., Christmann A., Gortner L., and Meyer S. Dravet syndrome: a new causative SCN1A mutation? // Clin. Case Rep. -2017. - Vol. 5, N. 5. - P. 613-615.
- Shi X., Yasumoto S., Nakagawa E., Fukasawa T., Uchiya S., Hirose S. Missense mutation of the sodium channel gene SCN2A causes Dravet syndrome // Brain Dev. - 2009. - Vol.31. - P. 758-762.
- Damaj L., Lupien-Meilleur A., Lortie A., Riou E., Ospina L., Gagnon L., Vanasse C. and Rossignol E. CACNA1A haploinsufficiency causes cognitive impairment, autism and epileptic encephalopathy with mild cerebellar symptoms // Eur J Hum Genet. - 2015. - Vol. 23(11) - P.1505-1512. doi: 10.1038/ejhg.2015.21.
- Lim C.X., Ricos M.G., Dibbens L.M., Heron S.E. KCNT1 mutations in seizure disorders: the phenotypic spectrum and functional effects // J Med Genet. - 2016. - Vol.53(4). - P.217-225. doi: 10.1136/jmedgenet-2015-103508.
- Vadlamudi L., Milne R.L., Lawrence K., Heron S.E., Eckhaus J., Keay D., Connellan M., Torn-Broers Y., Howell R.A., Mulley J.C., Scheffer I.E., Dibbens L.M., Hopper J.L., Berkovic S.F. Genetics of epilepsy: The testimony of twins in the molecular era // Neurology. - 2014. - Vol. 83, № 12. - P. 1042-1048. doi: 10.1212/WNL.0000000000000790.
- Ortrud K. Steinlein Genetics and epilepsy // Dialogues Clin Neurosci. - 2008. - Vol.10(1). - P. 29-38.
- Scheffer IE, Zhang Y-H, Jansen FE, Dibbens L. Dravet syndrome or genetic (generalized) epilepsy with febrile seizures plus? // Brain Dev. - 2009. - Vol. 31. - P. 394-400. doi: 10.1016/j.braindev.2009.01.001.
- Mullen SA, Scheffer IE. Translational research in epilepsy genetics. Sodium channels in man to interneuronopathy in mouse // Arch Neurol. - 2009. - Vol.66. - P.21-26. doi: 10.1001/archneurol.2008.559.
- Ottman R, Hirose S, Jain S, Lerche H, Lopes-Cendes I, Noebels JL, Serratosa J, Zara F, Scheffer IE. Genetic testing in the epilepsies - report of the ILAE genetics commission // Epilepsia. - 2010. - Vol. 51(4). - P.655-670.
- Lim CX, Ricos MG, Dibbens LM, Heron SE. KCNT1 mutations in seizure disorders: the phenotypic spectrum and functional effects // J Med Genet.- 2016. - Vol.53(4). - P.217-225. doi: 10.1136/jmedgenet-2015-103508.
- Villa C. and Combi R. Potassium channels and human epileptic phenotypes: an updated overview // Front Cell Neurosci. - 2016. - Vol. 10. - P. 81-93. doi: 10.3389/fncel.2016.00081.