В настоящее время достаточно хорошо изучена роль таких факторов риска развития ишемического инсульта, как артериальная гипертензия, атеросклероз, нарушение ритма сердца, инфаркт, курение, сахарный диабет, нарушение липидного обмена, изменения в системе гемостаза, применение оральных контрацептивов, злоупотребление алкоголем и др. [8]. Известно, что тяжесть ишемического инсульта возрастает при сочетании нескольких факторов риска, среди которых значительными являются артериальная гипертензия, гиперхолестеринемия, увеличение уровня липопротеинов низкой плотности, курение [37,49]. Внедрение в клиническую практику рациональной системы профилактики и лечения артериальной гипертензии, гиполипидемической терапии, эффективной эндартериоэктомии и стентирования брахиоцефальных артерий позволило существенным образом снизить частоту возникновения мозговых катастроф. Вместе с тем данные последних лет показали, что у пациентов с высоким теоретическим индивидуальным риском инсульта фактическая частота его возникновения существенным образом разнится *25+. Все это приводит к обсуждению влияния наследственной предрасположенности и механизмов, лежащих в основе повышения индивидуального риска развития ишемического инсульта [5].
Понятие о генах-кандидатах
Благодаря открытию и внедрению в практику метода полимеразной цепной реакции, развития технологии рекомбинантных ДНК, стало возможным выявление генов- кандидатов, белковые продукты которых могут хотя бы потенциально участвовать в развитии инсульта[35].
Гены, принимающие участие в развитии ишемического инсульта, подразделены на две категории: 1) увеличивающие риск развития ишемического инсульта, 2) влияющие на сосудистую реактивность, устойчивость ткани мозга к ишемии, размер очага. Эти категории не являются взаимоисключающими *20+. Определение роли конкретного гена в развитии ишемического инсульта является сложной задачей. Это связано с взаимодействием гена с другими факторами риска (артериальная гипертензия, сахарный диабет, повышение уровня фибриногена, нарушение липидного обмена) или модулированием их эффекта, а также с так называемым эффектом генной дозы. Эффект генной дозы - увеличение риска болезни при дефекте определенного гена в совокупности с другими. Часто такие комбинации являются синергическими, увеличивающими риск развития ишемического инсульта *12+. Кроме того, каждому подтипу ишемического инсульта соответствует дефект определенного гена, свои этиология и патогенез. В настоящее время лишь в единичных работах учитывается подтип ишемического инсульта. Наиболее изученной моделью для изучения наследственной предрасположенности в развитии инфаркта мозга является атеротромботический инсульт *6+. Кроме того, риск развития ишемического инсульта увеличивается не только под влиянием полиморфизма с участием одной пары нуклеотидов, но и при сочетании аллелей нескольких генов, т.е. имеет полигенную наследственную предрасположенность.
Имеется множество генов, определенные аллели которых ассоциируются с повышенным риском развития цереброваскулярных заболеваний. Так, широко изучаются полиморфные маркеры, относящиеся к генам системы гемостаза (к генам ФБ, тромбоцитарного гликопротеина, V, VIII и XII факторов свертывания, протромбина, тромбомодулина, генов, белков, участвующих в фибринолизе (tPA, PAI-1), ренин- ангиотензиновой системы, NO-синтетазы, липидов крови и гомоцистеина) *9, 20, 27, 28, 39+.
Патогенез тромбообразования у больных ишемическим инсультом
Не вызывает сомнения, что одной из основных причин возникновения ишемических инсультов является тромбоз церебральных артерий: до 50% острых нарушений мозгового кровообращения по ишемическому типу являются тромботическим или эмболическим осложнением атеросклеротического процесса в артериях крупного и среднего калибра *47+. Известно, что процесс тромбообразования зависит от множества факторов: гемодинамических, состояния сосудисто-тромбоцитарного и плазменного компонентов системы гемостаза, стадии развития атеросклеротической бляшки *44+. Р. Вирховым было выделено три фактора, предрасполагающих к развитию тромбозов: 1) нарушение тока крови; 2) повреждение стенки сосуда; 3) изменение реологических свойств крови. Компоненты триады являются лишь относительно самостоятельными и их значимость в патогенезе венозных и артериальных тромбозов неодинакова: ведущие причины развития венозных тромбозов - стаз и дефицит компонентов системы противосвертывания, артериальных - нарушение структуры сосудистой стенки и активация тромбоцитов *2+. Артериальные тромбы состоят в основном из тромбоцитов с небольшим содержанием фибрина и эритроцитов, поэтому их называют «белыми» тромбами в отличие от венозных - «красных» тромбов, состоящих преимущественно из эритроцитов и фибриновых нитей *23+. Различие венозных и артериальных тромбов не является абсолютным. Состав тромба определяется его возрастом. Так, тромбы, обнаруживаемые в церебральных и коронарных артериях при инфарктах, по составу преимущественно «белые» [14, 18].
Нормальное функционирование системы гемостаза обеспечивается сложными взаимодействиями компонентов плазмы, клеток крови и стенки сосудов, которые должны способствовать сохранению жидкого состояния крови в пределах кровеносных сосудов и быстрому тромбированию их поврежденных участков для предотвращения кровоизлияния в ткань. В норме адгезии тромбоцитов к неповрежденному эндотелию не происходит. Начальные стадии бляшки связаны с накоплением липидов в макрофагах с пролиферацией гладкомышечных клеток и образованием коллагена. Вследствие этих процессов образуется атеросклеротическая бляшка с ядром, в котором содержатся внеклеточные липиды. адро заключено в фиброзно-коллагеновую капсулу. Помимо свободного холестерина, в нем содержатся богатые липидами макрофаги, которые разрушаются, и их липидное содержимое увеличивает ядро. При разрывах мелких бляшек тромботические массы проникают внутрь бляшки, происходит стимуляция пролиферации гладкомышечных клеток и дальнейший рост бляшки. Когда она увеличивается настолько, что артерия теряет способность к ремоделированию, возникает гемодинамический стеноз церебральной артерии. Данные механизмы, лежащие в основе развития хронических стенозов, могут приводить к возникновению транзиторных ишемических атак в определенном сосудистом бассейне *3+.
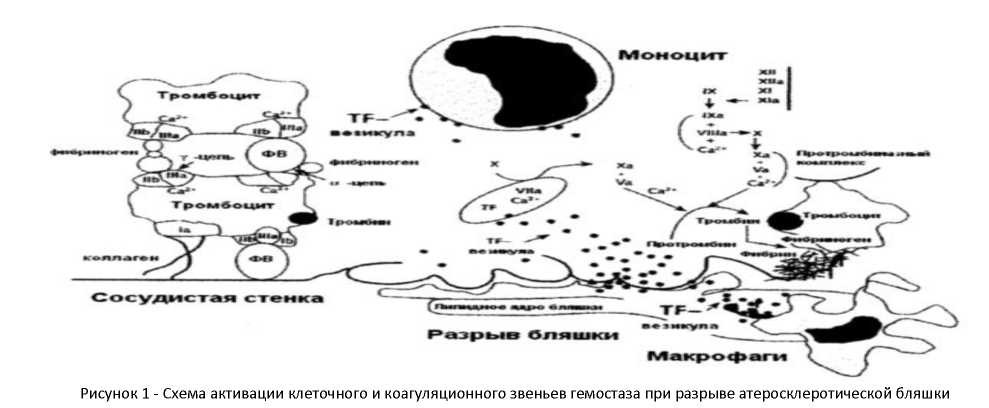
Второй возможный вариант развития процесса - быстрое образование тромба на бляшке, которое начинается либо по причине истончения и повреждения эндотелия, покрывающего бляшку, либо вследствие разрыва покрышки бляшки. При этом происходит снижение отрицательного заряда эндотелия и возникновение условий для формирования пристеночных тромбов из агрегатов тромбоцитов и фибрина (рис. 1). Это связано с тем, что активированные в результате контакта с коллагеном поврежденной сосудистой стенки тромбоциты выделяют коллагеназу и эластазу, расщепляющие соответствующие структурные белки, входящие в ее состав *18+. Помимо этого, тромбоциты высвобождают фактор Виллебранда, серотонин, АДФ, продукты превращения арахидоновой кислоты (простагландин PGI 2, тромбоксан А2). Эти биологические активные вещества вызывают вазоспазм, повышение проницаемости эндотелия, отек, усугубляя повреждение стенки сосуда, что в свою очередь усиливает адгезию и агрегацию тромбоцитов с образованием пристеночных тромбов *3, 19+.
Разрыв бляшек в местах выраженных стенозов сонных артерий может не сказываться на церебральном кровотоке, так как длительно существующий стеноз в сонной артерии способствует развитию коллатерального кровообращения. С другой стороны, разрыв бляшек, умеренно стенозирующих церебральные артерии, чаще проявляется симптомами острой недостаточности мозгового кровообращения из-за отсутствия развитого коллатерального русла *7+.
При возникновении инфаркта мозга большая роль отводится изменениям мозгового и магистрального кровотока артерий головы и шеи с формированием стенозов, окклюзий, патологической извитости. При этом причиной ишемии мозга является ограничение кровотока в атеросклеротически суженных сосудах на фоне временного изменения системного артериального давления под влиянием экстрацеребральных факторов, приводящих к возникновению зон так называемого турбулентного типа кровотока, являющегося одним из условий оседания форменных элементов, в первую очередь тромбоцитов. Это сопровождается повреждением сосудистой стенки, изменением реологических свойств крови, способствуя развитию тромботического ишемического инсульта *1, 19+.
Приблизительно у 40% больных развитие инсульта происходит по механизму кардиогенной и артериальной эмболии из распадающихся атером, пристеночных тромбов дуги аорты, магистральных артерий, внутрисердечных тромбов в результате аритмий, эндокардита, ревматизма и др. *4+.
Роль фибриногена в тромбообразовании у больных с инфарктом мозга
У больных с ишемической болезнью мозга, в том числе у перенесших ишемический инсульт, имеются изменения тромбоцитарного звена гемостаза. В процессах тромбообразования, в основе которых лежат адгезия и агрегация тромбоцитов, большая роль принадлежит фибриногену (один из факторов свертывания крови, который под действием тромбина превращается в нерастворимый фибрин, составляющий структурную основу тромба) *50+. Именно с ним взаимодействуют тромбоциты в области повреждения эндотелия. Фибриноген, иммобилизованный на поверхности эндотелиальных клеток, выполняет роль мостиков, связывающих активированные тромбоциты между собой и с коллагеном субэндотелиальных слоев, взаимодействуя с гликопротеином - специфическим рецептором мембраны тромбоцитов *22, 33, 36, 46+. Известно, что формирование тромбоцитарных агрегатов происходит с помощью этих рецепторова, способных взаимодействовать не только с фибриногеном, но и с фактором Виллебранда, фибронектином и витронектином. Активация GP является ключевым процессом, запускающим агрегацию тромбоцитов *3+. Таким образом, увеличение уровня фибриногена имеет определенное значение в развитии тромботических осложнений *15+.
В международных исследованиях PROCAM (the Prospective Cardiovascular Munster), PRIME (the Prospective Epidemiological Study of Myocardial Infarction), Framingham study, Northwick Park Heart было установлено, что повышение уровня фибриногена увеличивает в несколько раз риск развития инфарктов мозга и сердца *21, 31, 38+. Механизмы, лежащие в основе связи между повышением уровня фибриногена и риском ишемического инсульта, недостаточно ясны. Это может быть обусловлено тем, что фибриноген повышает вязкость крови, агрегацию тромбоцитов и эритроцитов, стимулирует пролиферацию эндотелиальных и гладкомышечных клеток, обеспечивая матрикс для роста клеток, задерживая тромбин, обладающий митогенной активностью *17+. Кроме того, накапливаясь в области атеросклеротической бляшки, фибрин стабилизирует тромбоцитарные агрегаты *11, 42, 45+.
Некоторые исследователи рассматривают фибриноген как маркер повреждения эндотелия *17+. Считается, что уровень фибриногена является чувствительным и одним из самых ранних факторов, отражающих повреждение эндотелиальных клеток. Определение его концентрации в сыворотке крови имеет значение для оценки тяжести и распространенности повреждения сосудов. Увеличение уровня фибриногена является прогностически неблагоприятным фактором, ассоциированным с увеличением риска смерти у больных с атеросклеротическим поражением сосудов головного мозга и сердца *20, 45+.
Гены фибриногена находятся на длинном плече 4-й хромосомы. Последовательность каждой из цепей фибриногена кодируется своим геном, однако значительная гомология свидетельствует об их возникновении в результате дупликации одного гена- предшественника *51+.
Фибриноген состоит из трех пар неидентичных полипептидных цепей, обозначаемых α, β и γ (рис. 2), является симметричной, вытянутой, слегка изогнутой молекулой размером 7x48 нм. N- концевые части всех трех полипептидов образуют центральную область взаимодействия двух половин молекулы фибриногена, которые ковалентно связаны между собой тремя дисульфидными мостиками, далее следует область, в которой все три субъединицы закручены в суперспираль, примерно по середине которой имеется короткая область нарушения регулярности структуры, являющаяся одним из участков специфического расщепления плазмином. За суперспиралью каждая из полипептидных цепей фибриногена образует свою структуру (С-концевые фрагменты) *3, 9+. Пространственная организация С-концевых фрагментов ß- и ү-цепей во многом сходна. Они образуют на концах молекулы фибриногена глобулы, латерально смещенные от оси суперспирали. За счет наличия S-S связи в ß-цепи ее концевая глобула несколько сдвинута к центру молекулы. Глобулярные участки ß- и ү-цепей вместе с дистальной частью суперспирали составляют D- фрагмент фибриногена. Согласно модели J. Weisel и соавт. после спирального участка ɑ-цепи загибаются, и их С-концы взаимодействуют друг с другом вблизи центра молекул. α- и ß- Цепи фибриногена синтезируются как единственные продукты соответствующих генов и состоят из 610 и 461 аминокислотных остатков соответственно. Фибриноген, содержащий удлиненную ү-цепь, менее эффективно взаимодействует с тромбоцитами, снижая их агрегационную активность *48+.
Фровень фибриногена плазмы крови зависит от многих факторов (возраст, пол, низкая физическая активность, артериальная гипертензия, курение, инсулинорезистентность) *13, 26, 29, 40+.
Фибриноген является белком острой фазы, его уровень в плазме повышается также при воспалении, инфекциях, травме и стрессе. Регуляция синтеза фибриногена осуществляется на уровне транскрипции. Синтез стимулируется гормонами, жирными кислотами, продуктами деградации фибриногена. Основным механизмом стимулирующего действия является секреция интерлейкина-6 макрофагами и моноцитами в ответ на фагоцитоз продуктов деградации фибриногена *35+.
Курение, вирусные инфекции, воспаление влияют на содержание фибриногена плазмы путем повышения количества лейкоцитов, секреции ими эластазы и интерлейкина-6 - основного стимулятора транскрипции ß-фибриногена в печени *16+. Прекращение курения приводит к некоторому снижению содержания фибриногена, но его уровень все же остается выше, чем у людей, никогда не куривших *41, 43+. Фровень фибриногена плазмы повышается при увеличении уровня глюкозы крови и инсулина *24+. Сильная положительная связь обнаружена между уровнем фибриногена, индексом массы тела и абдоминальным типом ожирения *10,32+. С возрастом содержание фибриногена также увеличивается. Ф женщин уровень фибриногена сыворотки крови выше, чем у мужчин, и более заметно его увеличение с возрастом. Прием эстрогенов приводит к снижению фибриногена, с этим хорошо согласуются данные об увеличении уровня в менопаузе *30+. Обнаружена положительная связь фибриногена с липопротеидами низкой плотности и триглицеридами, хотя зависимости между его концентрацией и липидным профилем крови выявлено не было [32].
СПИСОК ЛИТЕРАТУРЫ
- Боголепов Н.К. // Церебральные кризы и инсульт. - М: Медицина, 1972. – 392 c.
- Гусев Е.И., Скворцова В.И. // Ишемия головного мозга. - М: Медицина, 2001. – 7c.
- Панченко Е.П., Добровольский А.Б. Тромбозы в кардиологии. // Механизмы развития и возможности терапии. - М: Спорт и культура 1999. – C. 55—59.
- Шмидт Е.В., Смирнов В.Е. Вопросы эпидемиологии сосудистых заболеваний головного мозга. М: Медицина 1972.
- Aalto-Setala К., Palomaki Н., Miettinert Н. et al. Genetic risk factors and ischaemic cerebrovascular disease: role of common variation of the genes encoding apolipoproteins and angiotensin-converting enzyme. Ann Med 1998;30:224—233.
- Adams H.P.Jr., Bendixen B.H., Kappelle L.J. et al.Classification of subtype of acute ischemic stroke. Definitions for use in a multicenter clinical trial. TOAST. Trial of Org 10172 in Acute Stroke Treatment. Stroke 1993;24:35—41.
- Bansal В.С., Sood A.K., Bansal C.B. Familial hyperlipidemia in stroke in the young. Stroke 1986;17:1142—1145.
- Balleisen L., Bailey J., Epping P.H. et al. Epidemiology study on factor VII, factor VIII and fibrinogen in an industrial population, I: baseline data on the relation to age, gender, body weight, smoking, alcohol, pill using and menopause. Thromb Haemost 1985;54: 721—723.
- Baumann R.E., Henschen A.H. Human fibrinogen polymorphic site analysis by restriction endonuclease digestion and allele-specific polymerase chain reaction amplification: identification of polymorphisms at positions A 312 and В 448. Blood 1993; 82: 2117—2124.
- Broderick J., Brott T., Kothari R. et al. The Greater Cincinnati/Northern Kentucky Stroke Study: preliminary first-ever and total incidence rates of stroke among blacks. Stroke 1998;29:415—421.
- Brown E.T., Fuller G.M. Detection of a complex that associates with the b-fibrinogen G-455-A polymorphism. Blood 1998;92:3286—3293.
- Celentano A., Mancini F.P., Crivaro M. et al. Cardiovascular risk factors, angiotensinconverting enzyme gene I/D polymorphism, and left ventricular mass in systemic hypertension. Am J Cardiol 1999;83:1196—1200.
- Collet J.P., Soria J., Mirshahi M. et al. Dusart syndrome: a new concept of the relationship between fibrin clot architecture and fibrin clot degradability: hypofibrinolysis related toan abnormal clot structure. Blood 1993;82:2462—2469.
- Davies M.J. The contribution of thrombosis to the clinical expression of coronary atherosclerosis. Thromb Res 1996;82:1—32.
- Ferraresi P., Marchetti G., Legnani C. et al. The heterozygous 20210 G/A prothrombin genotype is associated with early venous thrombophilias and is not increased in frequency in arterial disease. Arterioscler Thromb Vasc Biol 1997;17:2418.
- Fishman D., Faulds G, Jeffery R et.al. The effect of novel polymorphisms in tl interleukin-6 (IL-6) gene on IL-6 transcription and plasma IL-6 levels, and an association with systemic-onset juvenile chronic arthritis. J Clin Invest 1998;102:1369—1376.
- Folsom A.R. Hemostatic risk factors for atherothrombotic disease: an epidemiologic view. Thromb Haemost 2001;86:366—373.
- Fuster V., Badimon L., Badimon J.J., Chesebro J.H. The pathogenesis of coronary artery disease and the acute coronary syndromes (1). N Engl J Med 1992;326:242—250.
- Gutstein D.E., Fuster V. Pathophysiology and clinical significance of atherosclerotic plaque rupture. Cardiovasc Res 1999;41:323—333.
- Hassan A., Hugh S. Markus. Genetics and ischaemic stroke. Brain 2000;123:1784—1796.
- Heinrich J., Balleisen L., Schulte H. et al. Fibrinogen and factor VII in the prediction of coronary risk: results from the PROCAM study in healthy men. Arterioscler Thromb 1994;14:54—59.
- Jang Y., Lincoff A.M., Plow E.F., Topol E.J. Cell adhesion molecules in coronary artery disease. J Am Coll Cardiol 1994;24:1591—1601.
- Jates P.O. A change in the pattern of cerebrovascular disease. Lancet 1964;1:65—69.
- Kannel W.B., Wilson P.R., Bleanger A.J., Gargon D.R. Diabetes, fibrinogen, and risk of cardiovascular disease. The Framingam experience. Am Heart J 1990;120:87—94.
- Kokubo Y., Chowdhury A.H., Date C. Age-dependent association of apolipoprotein E genotype with stroke subtypes in a Japanese rural population. Stroke 2000;31:6:1299—1309.
- Krobot K., Hense H.W., Cremer P. et al. Determinants of plasma fibrinogen: relation of body weight, waist-to-hip ratio, smoking, alcohol, age, and sex: results from the second MONICA Augsburg Survey 1989—1990. Arterioscler Thromb 1992;12:780—788.
- Lane D., Grant P. Role of hemostatic gene polymorphisms in venous and arterial thrombotic disease. Blood 2000;95:1517—1532.
- Margaglione M., Seripa D., Gravina C. et al. Prevalence of apolipoprotein E alleles in healthy subjects and survivors of ischemic stroke: an Italian case-control study. Stroke 1998;29:399—403.
- Meade T.W., Brozovic M., Chakrabarth R. et al. An epidemiological study of the haemostatic and other effects of oral contraceptives. Br J Haematol 1976;34:353—364.
- Meade T.W., Haines A.P., North W.R. Characteristics affecting fibrinolytic activity and plasma fibrinogen concentration. Br Med J 1979;1.
- Meade T.W., Mellows S., Brozovic M. et al. Haemostatic function and ischaemic heart disease: principal results of the Northwick Park Heart Study. Lancet 1986;2:533—537.
- Moller L., Kristensen T.S. Plasma fibrinogen and ischemic heart desease risk factor. Arterioscler Tromb 1991;11:145—157.
- Peerschke E.I. Stabilization of platelet-fibrinogen interactions is anintegral property of the glycoprotein IIb—IIIa complex. J Lab Clin Med 1994;124:439—446.
- Pohjasvaara T., Mantyla R., Salonen О. et al. How complex interactions of ischemic brain infarcts, white matter lesions, and atrophy relate to poststroke dementia. Arch Neurol 2000;57:1295—1300.
- Princen U.M.G., Nieuwenhuizen W., Yap S.H. Direct evidence of transcriptional control of fibrinogen and albumin synthesis in rat liver during the acute phase response. Biochem Biophys Res Commun 1981;102:109—116.
- Savage B., Saldivar E., Ruggeri Z.M. Initiation of platelet adhesion by arrest onto fibrinogen or translocation on von Willebrand factor. Cell 1996;84:289—297.
- Scarabin P.Y., Bara L., Ricard S. et al. Genetic variation at the
- b-fibrinogen locus in relation to fibrinogen concentrations and risk of myocardial infarction: the ECTIM study. Arterioscler Thromb 1993;13:886—891.
- Scarabin P.Y., Arveiler D., Amouyel P. et al. Plasma fibrinogen explains much of the difference in risk of coronary heart disease between France and Northern Ireland. The PRIME study. Atherosclerosis 2003;166:103—109.
- Schmidt H., Schmidt R., Niederkorn K. et al. b-fibrinogen gene polymorphism (C148 to T) is associated with carotid atherosclerosis. Arterioscler Thromb Vasc Biol 1998;18:487.
- Sechi L.A., Zingaro L., Catena C. et al. Relationship of fibrinogen levels and hemostatic abnormalities with organ damage in hypertension. Hypertension 2000;36:978—991.
- Sharma P., Carter M.L., Barley J., Brown M.M. Molecular approach to assessing the genetic risk of cerebral infarction. J Hum Hypertens 1994;8:645—648.
- Smith E.B. Fibrinogen, fibrin and the arterial wall. Eur Heart J 1995;16:24—33.
- Staessen J.A., Fagard R., Thijs L. et al. Randomised double-blind comparison of placebo and active treatment for older patients with isolated systolic hypertension: the Systolic Hypertension in Europe (Syst-Eur) Trial Investigators. Lancet 1997;350:757—764.
- Tracy R.P. Epidemiological evidence for inflammation in cardiovascular disease. Thromb Haemost 1999;82:826—831.
- Tybjaerg H.A., Agerholm L.B., Humphries S.E. et al. A common mutation (G-455A) in the beta-fibrinogen promoter is an independent predictor of plasma fibrinogen, but not of ischemic heart disease. A study of 9,127 individuals based on the Copenhagen City Heart Study. J Clin Invest 1997;99:3034—3039.
- van der Bom J.G., Bots M.L., Haverkate F. et al. Reduced response to activated protein C is associated with increased risk for cerebrovascular disease. Ann Intern Med 1996;125:265—269.
- Wei X., Fu Y., Ni P.H., Song Y.Y., Chen S.D. The relationship between the five beta-fibrinogen gene polymorphisms and cerebral infarction. Abstract 2005; Dec: 44(12): 914—917. (TIMP-1) increased gene expression following focal stroke. Stroke 1998;29:516—520.
- Weisel J. W, Stavffacher C. V, Bullitt E., Cohen C.A. A model of fibrinogen: domans and seguence. Science 1985;230:1388—1391.
- Wolf P.A., D'Agostino R.B., O'Neal M.A. et al. Secular trends in stroke incidence and mortality: the Framingham Study. Stroke 1992; 23:1551— 1555.
- Woodward M., Low G.D.O., Rumley A. Fibrinogen as a risk factor for coronary heart disease and mortality in middle-aged men and women. Eur Heart J 1998;19.
- Yu S., Sher В., Kudryk В., Redman C.M. Fibrinogen precursors. Order of assembly of fibrinogen chain. J Biol Chem 1984;259:47—59.