АННОТАЦИЯ
Цель работы: изучить особенности клинического полиморфизма прогрессирующей мышечной дистрофии Дюшенна/Беккера среди пациентов с нервно-мышечными заболеваниями в Узбекистане. Ретроспективно и проспективно были проанализированы 106 пациентов мужского пола с псевдогипертрофическими формами прогрессирующих мышечных дистрофий: 93 больных с ПМДД в возрасте от 3 лет до 18 лет и 13 - с ПМДБ в возрасте от 10 лет до 25 лет, находящихся под наблюдением в отделении медико-генетического консультирования Республиканского центра «Скрининг матери и ребёнка» (г. Ташкент) в период с 2004 по 2014 гг. Выводы. В 27,7% случаях выявленные родословные с отчётливо прослеживающими заболеваниями в семье. Установлены корреляционные взаимосвязи клинических и биохимических показателей со степенью поражения мышечного аппарата, в тоже время уровень КФК в сыворотке крови отражал уровень двигательных нарушений только в терминальных стадиях заболевания и не являлся маркёром эффективности проводимых лечебных мероприятий. У больных с данной патологией характерные изменения электромиографических показателей и выраженность спонтанной активности отражает характер и степень остроты процесса, а также возможность прогноза качества жизни и исхода заболевания
Ключевые слова: прогрессирующие мышечные дистрофии, мышечная дистрофия Дюшенна, клиника, диагностика, дети
Введение. Частыми наследственными заболеваниями условно принято считать такие, распространенность которых составляет более 1:50000 населения (Гинтер Е.К., Зинченко Р.А., 2006). К ним относятся такие как миотоническая дистрофия, хорея Гентингтона, миодистрофия Дюшенна/Беккера, моторно-сенсорные нейропатии.
В Республике Узбекистане реализуется Государственная программа «Скрининг матери и ребенка», принятая Постановлением Кабинета Министров №140 от 08.04.98 г. по созданию государственной системы раннего выявления врождённых и наследственных заболеваний путём проведения массового неонатального скрининга новорожденных и дородового обследования беременных группы риска. Особую актуальность приобрели работы по анализу генов прогрессирующих миодистрофий (ПМД) в связи с их значимостью для решения многих проблем оказания помощи пациентам, страдающим от этих весьма тяжелых и инвалидизирующих наследственных болезней. В первую очередь внимание естественно обращено на наиболее частые и клинически важные формы ПМД, среди которых миодистрофии Дюшенна/Беккера и Эрба-Рота [1, 3, 4, 5 ].
Учитывая высокую частоту заболевания, тяжёлую степень инвалидизации и раннюю смертность пациентов с прогрессирующей мышечной дистрофии Дюшенна/Беккера (ПМДД/Б) актуальным и своевременным является изучение клинического полиморфизма данного заболевания в нашем регионе для создания алгоритма дифференциальной диагностики и регистра больных с целью предупреждения рождения детей с дефектом гена дистрофина в семьях, отягощённых по ПМДД/Б.
Цель исследования: Анализ особенностей клинического полиморфизма
прогрессирующей мышечной дистрофии Дюшенна/Беккера среди пациентов с нервномышечными заболеваниями в Узбекистане.
Материал и методы исследования: Ретроспективно и проспективно были проанализированы 106 пациентов мужского пола с псевдогипертрофическими формами прогрессирующих мышечных дистрофий: 93 больных с ПМДД в возрасте от 3 лет до 18 лет и 13 - с ПМДБ в возрасте от 10 лет до 25 лет, находящихся под наблюдением в отделении медикогенетического консультирования Республиканского центра «Скрининг матери и ребёнка» (г.
Ташкент) в период с 2004 по 2014 гг. Диагноз установлен на основании клинических проявлений заболевания, биохимических исследований (определение уровня активности
креатинфосфокиназы (КФК) и лактатдегидрогеназы (ЛДГ) в сыворотке крови) а также проводилась электронейромиография с использованием концентрических игольчатых электродов. Оценка двигательных функций проводилась с использование шкалы MRS (Modified Rankin Scale).
Результаты и обсуждения: Генеалогический анализ выявил наличие Х-сцеплененного рецессивного наследования ПМДД/Б в 23 (27,7%) семьях, в 60 (72,3%) семьях - выявлены единственные случаи заболевания.
При клиническом осмотре группы больных с МДД/Б мышечная сила составила в: 5 баллов - 1,6 % (1); 4 балла - 49,2% (31); 3 балла - 30,1 %(19); 2 балла - 14,3% (9); 1 балл - 4,7% (3).
Так средний возраст у пациентов в зависимости от количества баллов по шкале MRS составил: в группе 5 баллов - 3 года; 4 балла - 5 лет 10 месяцев; 3 балла - 6 лет 9 месяцев; 2 балла - 12 лет 3 месяца; 1 балл - 15 лет 6 месяцев.
Уровень КФК при различной степени мышечного поражения составил: в группе 5 баллов - 5025 u/l; 4 балла - 4523 u/l; 3 балла - 5219 u/l; 2 балла - 3188 u/l; 1 балла - 1258 u/l (табл. 1).
Таблица 1- Показатели мышечной силы шкале MRS в зависимости от возраста и уровня КФК
|
Мышечная сила в баллах |
Количество больных |
Средний возраст больных |
Средний уровень КФК (u/l) в крови |
|
5 баллов |
1,6 % (1) |
3 года |
5025 |
|
4 балла |
49,2 % (31) |
5 лет 10 месяцев |
4523 |
|
3 балла |
30,1 % (19) |
6 лет 9 месяцев |
5219 |
|
2 балла |
14,3 % (9) |
12 лет 3 месяца |
3188 |
|
1 балл |
4,7 % (3) |
15 лет 6 месяцев |
1258 |
Одним из важных этапов в диагностике ПМДД/Б является электронейромиография с использованием концентрических игольчатых электродов (игольчатая ЭНМГ). Простой, высокоинформативный и широко доступный метод, который позволяет не только идентифицировать уровень поражения двигательной единицы, но и на основании ЭНМГ- мониторинга оценить степень выраженности патологического процесса на различных стадиях заболевания.
Нами было обследовано 32 больных с диагнозом ПМДД/Б в возрасте от 6,5 до 24 лет, из них 4 пациента с ПМД Беккера. У всех больных были жалобы на слабость конечностей разной степени, нарушение походки. Для уточнения характера поражения и уточнения степени денервации всем больным было проведено игольчатая ЭНМГ с применением концентрических игольчатых электродов на аппарате «Нейрон-Спектр- 4/ВПМ». ЭНМГ проводилась в наиболее атрофированных мышцах, а именно в верхней группе мышц нижних конечностей (передняя, латеральня и медиальная бедренные мышцы). В ходе обследования проводилось тест на выявления спонтанной активности в мышцах и определение ПДЕ (потенциалы двигательных единиц). Спонтанная активность в виде фибрилляций, фасцикуляций и положительных острых волн фиксировалась с последующей обработкой данных. Для выявления ПДЕ больному давалась команда сопротивляться против направленной силы, для минимальной активации обследуемого мышцы. Появившиеся потенциалы фиксировались с последующей обработкой. При анализе ПДЕ измерялось длительность, форма и амплитуда. Для выявления средних показателей регистрировалась 20 ПДЕ из разных участков обследуемой мышцы.
Так, у всех пациентов длительность ПДЕ составляло меньше 10 мс и по гистограмме соответствовало II стадии денервационно-реиннервационного процесса (ДРП) (рис.1). Во всех случаях форма ПДЕ изменена в виде полифазных кривых. У всех обследованных было отмечено снижение средней длительности ПДЕ: на 36,3% при ПМД Дюшенна и на 18,9% при ПМД Беккера, что соответствовало II стадии денервационно-реиннервационного процесса (ДРП).
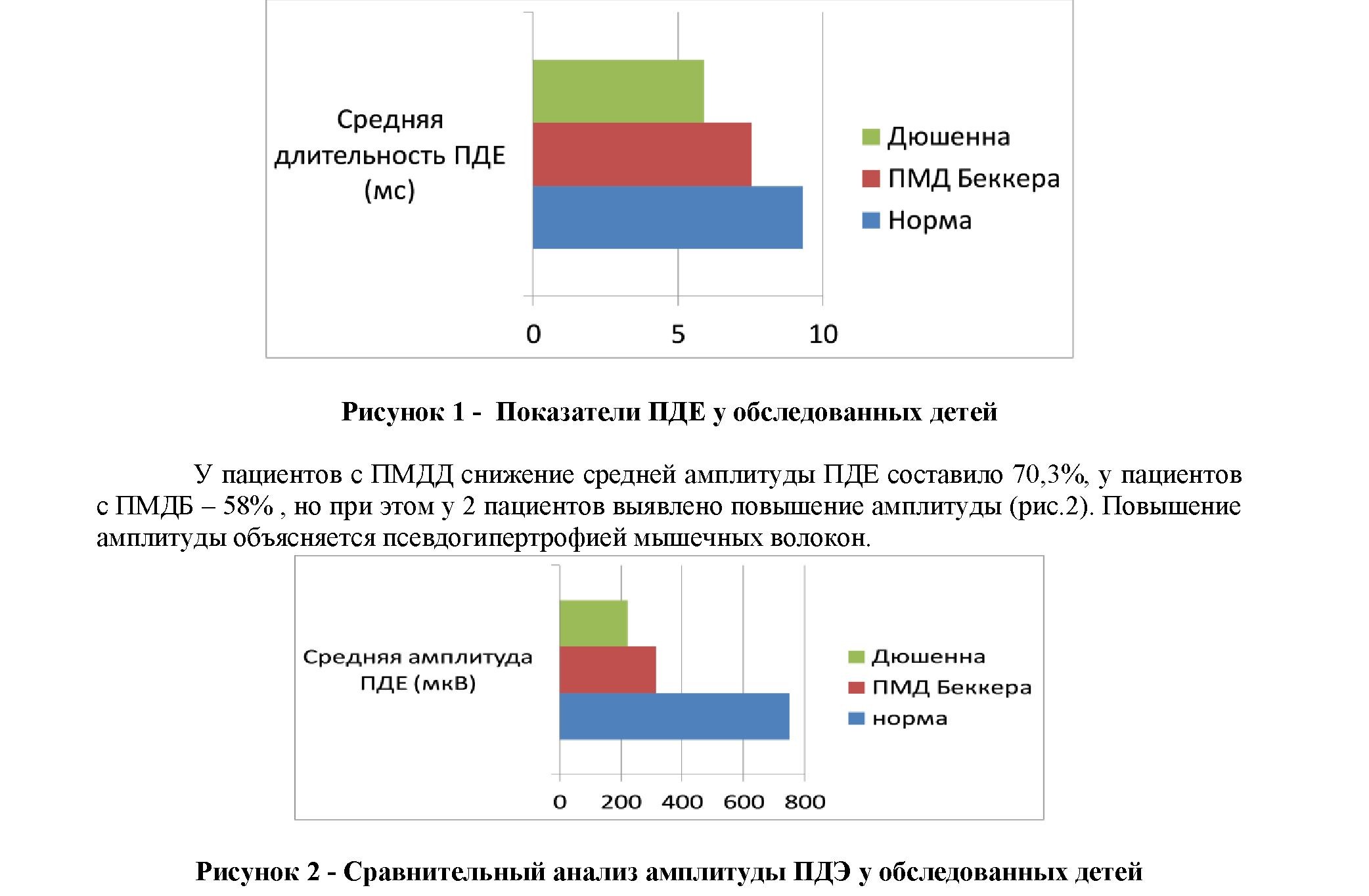
У 24 пациентов при обследовании наблюдалось спонтанная активность в виде потенциалов фибрилляций (ПФ), который варьировался по качеству и количеству. У 8 пациентов, с выраженными изменениями и быстрым прогрессированием спонтанная активность не выявлялось. Так, у пациентов с наиболее давним процессом (наиболее ранний возраст) он выявлялся в наименьшем количестве и низкой амплитуды, чем у пациентов с относительно недавним процессом. Также отмечалось спонтанная активность в виде положительных острых волн у 9 пациентов, при этом данный феномен наблюдалось у пациентов с наиболее давним процессом.
Выводы: По литературным данным приблизительно 30% всех случаев связаны с мутациями de novo в яйцеклетке матери больного, а остальные 70% — обусловлены гетерозиготностью матери по патологической мутации в гене дистрофина. Однако, родословные с отчётливо прослеживающими заболеваниями в семье, по нашим данным составили около 27,7 %.
Проанализировав данные клинических и биохимических обследований на момент первичного обращения была выявлена прямая корреляции только между возрастом и степенью поражения мышечного аппарата, в тоже время уровень КФК в сыворотке крови отражал уровень двигательных нарушений только в терминальных стадиях заболевания и не являлся маркёром эффективности проводимых лечебных мероприятий.
Исходя из данных электронейромиографии можно заключить, что у больных с ПМДД/Б характерные изменения ПДЕ и выраженность спонтанной активности отражает характер и степень остроты процесса, а также возможность прогноза качества жизни и исхода заболевания.
ЛИТЕРАТУРА
- Зинченко Р.А. Эпидемиология наследственных болезней в российских популяциях.// Автореф. дис.докт.мед.наук. - М. - 2001. - 46 с.
- Кириленко Н.Б. Особенности нозологического спектра и клинических характеристик наследственных болезней нервной системы в городах Волгоград и Волжский // Автореф.дис.канд.мед.наук. - М. - 2005. - 26 с.
- Корень О.Л. Распространенность и клинический полиморфизм наследственных заболеваний с поражением нервной системы в г.Новокузнецке: Автореф.дис.канд.мед.наук. - М. - 2005. - 24 с.
- Крахмалева И.Н., Липатова Н., Шишкин С.С., Шаховская Н.И. Структура дистрофинового гена у больных миодистрофией Дюшенна. // Журн. невропатолог и психиатр. - 1999. - №3. - С. 41.
- Рахмонов Р.А. Наследственные болезни нервной системы в Таджикистане: Автореф.дис.докт.мед.наук. - М., 2004. - 44 с.