Ишемический инсульт является важнейшей глобальной медицинской и социальной проблемой, т.к. его последствия обуславливают высокий уровень смертности и инвалидизации. Наряду с вопросами лечения и реабилитации больных, актуальным является вопрос профилактики заболевания, которая подразумевает под собой отбор группы лиц, имеющих высокий риск возникновения ишемического инсульта. Особую группу риска составляют пациенты с метаболическим синдромом, поскольку они сочетают в себе многие предрасполагающие факторы развития сердечно-сосудистых заболеваний.
Целью этой обзорной статьи является анализ факторов риска и этиопатогенетических причин развития острого нарушения мозгового кровообращения у пациентов с метаболическим синдромом.
Введение. В современном мире одно из ведущих мест среди причин смертности и инвалидизации населения принадлежит инсультам. Метаболический синдром (МС) рассматривается как предиктор развития сердечнососудистых заболеваний, в том числе и ишемического инсульта (ИИ). Пациенты с метаболическим синдромом имеют множество факторов риска: повышенное
артериальное давление (АД), инсуллинорезистентность или повышенный уровень глюкозы крови, абдоминальное ожирение, дислипидемии. Все эти факторы запускают и поддерживают формирование атеросклероза - одну из основных причин ишемического инсульта [1].
Результаты и обсуждение. Совместное влияние метаболических нарушений в настоящее время известных как метаболический синдром был впервые описан Kylin в 1920 как сочетание гипертонии, гипергликемии и подагры [2]. Vague отмечал, что ожирение по мужскому типу наиболее часто ассоциируется с диабетом и ССЗ [3]. В 1988 Reaven отметил клиническую важность МС и использовал термин «синдром X» [4].
В 1989 году Каплан назвал это сочетание «смертельный квартет», а также был применен термин «синдром инсулинорезистентности [5]. В настоящее время используется термин «метаболический синдром», который сочетает в себе многие предрасполагающие факторы развития сердечно-сосудистых заболеваний, такие как: повышенное АД, инсуллинорезистентность и/или повышенный уровень глюкозы крови, абдоминальное ожирение, дислипидемии. Зачастую у пациентов с МС повышен уровень мочевой кислоты, СРБ, изменен гормональный профиль. Все эти факторы запускают и поддерживают формирование атеросклероза- одной из основных причин сердечно - сосудистых заболеваний.
Экспертные группы пытались разработать единое определение МС. Наиболее широкие исследования были произведены Всемирной организацией здравоохранения (ВОЗ), Тhe European Group for the Study of Insulin Resistance и the National Cholesterol Education Program—Third Adult Treatment Panel (ATP III) [6,7,8].
Все группы признавали основные компоненты МС: ожирение, инсулинорезистентность, дислипидемия и артериальная гипертензия. Тем не менее, они описывали различные клинические критерии для определения установления МС. Например, в отличие от двух других определений, определение ATP III не требовало наличия нарушенной регуляции глюкозы или резистентности к инсулину в качестве основного компонента. В 2005 и (повторно в 2009) году организация IDF было разработала единый диагностический инструмент для определения МС. Было решено использовать определение ATP III 2001 года в качестве отправной точки. Несмотря на то, что резистентность к инсулину является важным компонентом МС, его измерение не является необходимым для нового определения, ввиду трудоемкости регулярного измерения в клинической практике, в то время как измерение абдоминального ожирения является доступным. В результате были разработаны критерии установления МС (Таблица 1). Для установления наличия МС пациент должен обладать абдоминальным ожирением и любыми двумя признаками из таблицы [8].
Таблица 1 - Критерии установления МС
|
Параметр |
Значение |
|
Центральное ожирение |
Обхват талии: этническая специфика муж > 94 см; жен > 80 см. |
|
Повышенные триглицериды |
> 150 мг/дл (1,7 ммоль/л) (или специфическое лечение этой липидной аномалии) |
|
Сниженный холестерин липопротеидов высокой плотности |
(или специфическое лечение этой липидной аномалии) |
|
Повышенное артериальное давление |
Систолическое: > 130мм рт.ст. Диастолическое: > 85мм рт.ст. (лечение ранее диагностированной гипертонии) |
|
Повышенный уровень глюкозы плазмы натощак |
Глюкоза плазмы натощак >100мг/дл (5,6 ммоль/л) или (ранее диагностированный 2 тип диабета) |
В дополнение к определению в МС, участники семинара IDF подчеркнули ряд других параметров, которые могут оказаться связаны с МС: эндотелиальная дисфункция, изменение концентраций биомаркеров жировой ткани лептина и адипонектина, инсулинорезистентность, протромботические состояния и т.д.
При МС происходят ряд метаболических изменений, которые описаны ниже.
Абдоминальное ожирение. Абдоминальное ожирение (АО) считается одним из важнейших факторов риска ССЗ, таких как артериальная гипертензия, ишемическая болезнь сердца, ОНМК. АО используется клиницистами как своеобразный маркер прогноза возникновения ССЗ.
Адипоциты (жировые клетки) действует как эндокринный орган, и играют существенную роль в патогенезе абдоминального ожирения [9]. Адипоциты синтезируют гормоноподобное вещество лептин, который контролирует потребление пищи и энергетический обмен. В случае ожирения синтез лептина изменяется, что приводит к неконтролируемому пищевому поведения. Систематическое воспаление у пациентов с АО приводит к повышенной концентрации СРБ. Как указано в работах P.J. Enriori, СРБ может играть роль в развитии резистентности к лептину, результатом чего является снижение энергетического обмена и неконтролируемый аппетит [10]. Увеличение концентрации СРБ и лептина были связаны с повышенным риском ССЗ. Лептин оказался независимым предиктором ССЗ, в то время как СРБ таким не являлся. По данным C.J. Lavie, увеличение маркеров воспаления связано с резистентностью к инсулину и ССЗ [11,12].
Помимо лептина адипоциты синтезируют фактор некроза опухолей-альфа, ингибитор активатора плазминогена-1, интерлейкин-6, ангиотензиноген, инсулиноподобного фактора роста-1. Некоторые из этих факторов, воздействуя на инсулинорецепторы, приводят к развитию инсулинорезистентности, в то время как другие запускают процесс повышения АД. В ответ на ИР на первом этапе развивается гиперинсулинемия [13].
Инсулин, помимо своих основных функций оказывает следующие эффекты [13]:
- приводит к гиперволемии путем повышенной реабсорбции натрия в проксимальных канальцах почек;
- повышает активность симпатоадреналовой системы, вызывая вазоконстрикцию и повышение общего периферического сосудистого сопротивления (ОПСС);
- стимулирует пролиферацию гладкомышечных клеток сосудов.
АО оказывает ряд побочных эффектов на гемодинамику, структуру и функции сердечно-сосудистой системы [13]. АО увеличивает общий объем крови, сердечный выброс и сердечную нагрузку. Пациенты с АО имеют высокий сердечный выброс, но низкий уровень общего периферического сопротивления при любых показателях артериального давления. Это увеличение сердечного выброса вызвано повышенной активностью симпатической нервной системы, что приводит к увеличению частоты сердечных сокращений. Гиперактивация симпатической нервной системы указывает на нейрогенный характер АГ при АО. При АО в следствии инсулинорезистентности происходит активация симпатоадреналовой системы, что приводит к активации ренин-ангиотензин-альдостероновой системы (РААС). ИР и активация РААС суммарно приводят к артериальной гипертензии, которая характеризуется отсутствием ночного снижения АД [13].
У людей с АО часто развивается дилатация левого желудочка из-за увеличения его наполнения и повышенного объема крови [14]. Не зависимо от артериального давления и возраста, ожирение увеличивает риск гипертрофии левого желудочка (ГЛЖ), а также других структурных аномалий, включая концентрическое ремоделирование и концентрическую гипертрофию левого желудочка [15]. АО приводит к дилатации левого предсердия, из-за увеличением объема циркулирующей крови и аномального заполнения во время диастолы. Кроме того у пациентов с АО имеется склонность к частым и сложным желудочковым аритмиям. Эти нарушения многократно увеличивают риск ССЗ, в том числе ишемического инсульта [16].
Многочисленные исследования выявили связь между ИМТ и инсультом. Показано, что увеличение ИМТ на 1 единицу, повышает риск развития ишемического инсульта на 4% и на 6% геморрагического инсульта. Такой рост риска может быть связан с высокой распространенностью артериальной гипертензии при АО и провоспалительного состояния, которые сопровождают избыточное накопление жировой ткани [17].
Гипергликемия. К обратимым метаболическим изменениям относят активацию полиолового пути обмена глюкозы, истощение запасов миоинозитола в клетках, активацию протеинкиназы С (повышенная активность этого фермента в условиях гипергликемии сопровождается активацией процессов перекисного окисления липидов, обладающих цитотоксическими эффектами), повышенное образование ранних продуктов гликозилирования и связанных с ними свободных радикалов. Необратимые метаболические изменения — это образование конечных необратимых продуктов гликозилирования и их связывание с компонентами внеклеточного матрикса и нуклеиновыми кислотами ядер клеток, изменение третичной структуры белков базальной мембраны, нарушение связывающей способности белков матрикса, повышение частоты генетических мутаций [18].
В условиях гипергликемии происходит неферментативное гликозилирование белков, которые в зависимости от периода их полужизни подразделяют на два класса: обратимые, обладающие наибольшим повреждающим действием, изменяющие структуру и метаболизм основных белков организма (коллагена, миелина, кристаллина, ДНК), взаимодействуют с атерогенными фракциями липидов (тем самым снижая их клиренс из кровотока и повышая атерогенность сыворотки крови), активируют процессы перекисного окисления липидов [18].
По данным Мак Дермотт М.Т. [19] конечные продукты гликозилирования (КПГ) самостоятельно способствуют развитию и поддержанию высокого артериального давления у больных сахарным диабетом, нарушая нормальную чувствительность стенок сосудов к действию сосудорасширяющих веществ. Необратимость молекул КПГ объясняет продолжающееся прогрессирование микро- и макрососудистых осложнений даже при очень хорошей компенсации инсулинорезистентности.
Ю. К. Ширяева в своих работах описалa, что конечные продукты гликозилирования, циркулирующие в кровеносном русле связываются сбазальной мембраной, эндотелием сосудов и рыхлой соединительной тканью, что приводит к формированию эндотелиальной дисфункции[20]. Большинство современных исследователей утверждают, что эндотелий играет центральную роль во многих патофизиологических процессах, приводящих к развитию микрососудистых осложнений и атеросклеротических изменений крупных сосудов. Уникальное положение эндотелиальных клеток на границе между циркулирующей кровью и тканями делает их основной мишенью в развитии ангиопатий [20].
Из всех многочисленных факторов, синтезируемых эндотелием, роль "модератора" всех функций эндотелия принадлежит эндотелиальному фактору релаксации, или оксиду азота (NO). Именно этот фактор регулирует активность и последовательность "запуска" всех остальных биологически активных веществ, продуцируемых эндотелием [20].
Нарушение NO-продуцирующей функции эндотелия в первую очередь связано с развитием ангиопатий и атерогенезом. Недостаточная продукция NO приводит не только к сниженной релаксации сосудов и их спазму, но и к повышенной проницаемости сосудов для белков и липопротеинов, к ускоренной пролиферации гладкомышечных клеток, к беспрепятственной экспрессии поверхности эндотелиальных тромбообразованию. Все эти к дисбалансу между адгезивных молекул на клеток, к повышенному процессы приводят сосудорасширяющимии антитромботическими и противовоспалительными сосудосуживающими, протромботическими, провоспалительными, антисклеротическими, пролиферативнымифакторами в сторону преобладания последних [21].
Как указано в работе [21] измененный эндотелий экспрессирует молекулы адгезии, в частности ICAM-1, облегчающие проникновение моноцитов, переполненных липидами, в субэндотелий. Эти нарушения способствуют аккумулированию липидов в сосудистой стенке, пролиферации и миграции в интиму артерий гладкомышечных клеток и усилению продукции ими коллагена и эластина, развитию микроагрегатов тромбоцитов. Последние формируют условия для микроэмболий vasavasorum крупных артериальных сосудов с локальными изменениями сосудистой стенки, что в конечном итоге и приводит к развитию атеросклероза и тромбозу.
Роль дислипидемий. Согласно мировой статистике у пациентов с МС наблюдается гипертриглицеридемия в сочетании с пониженным уровнем холестерина липопротеидов высокой плотности (ХС ЛВП). Концентрации других классов липопротеидов, как правило, повышены: липопротеидов очень низкой плотности (ЛОНП), липопротеидов промежуточной плотности (ЛПП) и хиломикрон. Что касается липопротеидов низкой плотности (ЛНП), то нередко их концентрация остается в пределах нормы, однако по своему фенотипу - это липопротеиды типа В, т.е. они имеют меньшие размеры и несколько большую плотность, чем ЛНП у здоровых лиц [22].
ЛНП В типа длительное время циркулируют в кровотоке, окисляются и захватываются макрофагами в сосудистой стенке. Хроническая гипергликемия способствует повышенной проницаемости окисленных ЛНП в сосудистую стенку, поскольку приводит к эндотелиальной дисфункции и как следствие к повышенной проницаемости сосудов. Эти процессы активируют высвобождение цитокинов (IL-I, TNFa), которые в свою очередь активируют процесс воспаления в стенке артерии [23]. Воспалительные реакции в стенке артерий являются еще одним патологическим звеном для образования ангиопатий и атеросклероза.
Роль гиперинсулинемии. По данным А.Ю. Рунихина [24] гиперинсулинемия напрямую стимулирует синтез триглицеридов из фруктозы в печени и снижает синтез фосфолипидов, что приводит к дефициту липопротеидов высокой плотности и образованию перечисленных выше патологических типов липопротеидов низкой плотности. Инсулинорезистентность и гиперинсулинемия являются также непосредственной причиной АГ.
Инсулинорезистентность приводит к повышению АД за счет стимуляции активности симпатической нервной системы, снижения синтеза оксида азота в эндотелии, активности Na, К-АТФазы в гладкомышечных клетках сосудов, что вызывает усиление поступления ионов кальция в эти клетки и их сокращение. Гиперинсулинемия также способствует возникновению АГ за счет усиления реабсорбции натрия и воды в почках, стимуляции неренинового пути синтеза ангиотензина II в тканях и за счет гипертрофии мышечной оболочки (медии) резистивных артерий. Не вызывает сомнения участие инсулинорезистентности (ИР) в формировании протромботического состояния. В большом количестве клинических исследований обнаружены четкие взаимоотношениямежду синдромом инсулинорезистентности и уровнем PAI-1, как у больных с МС так и убольных с изолированной инсулинорезистентности [25].
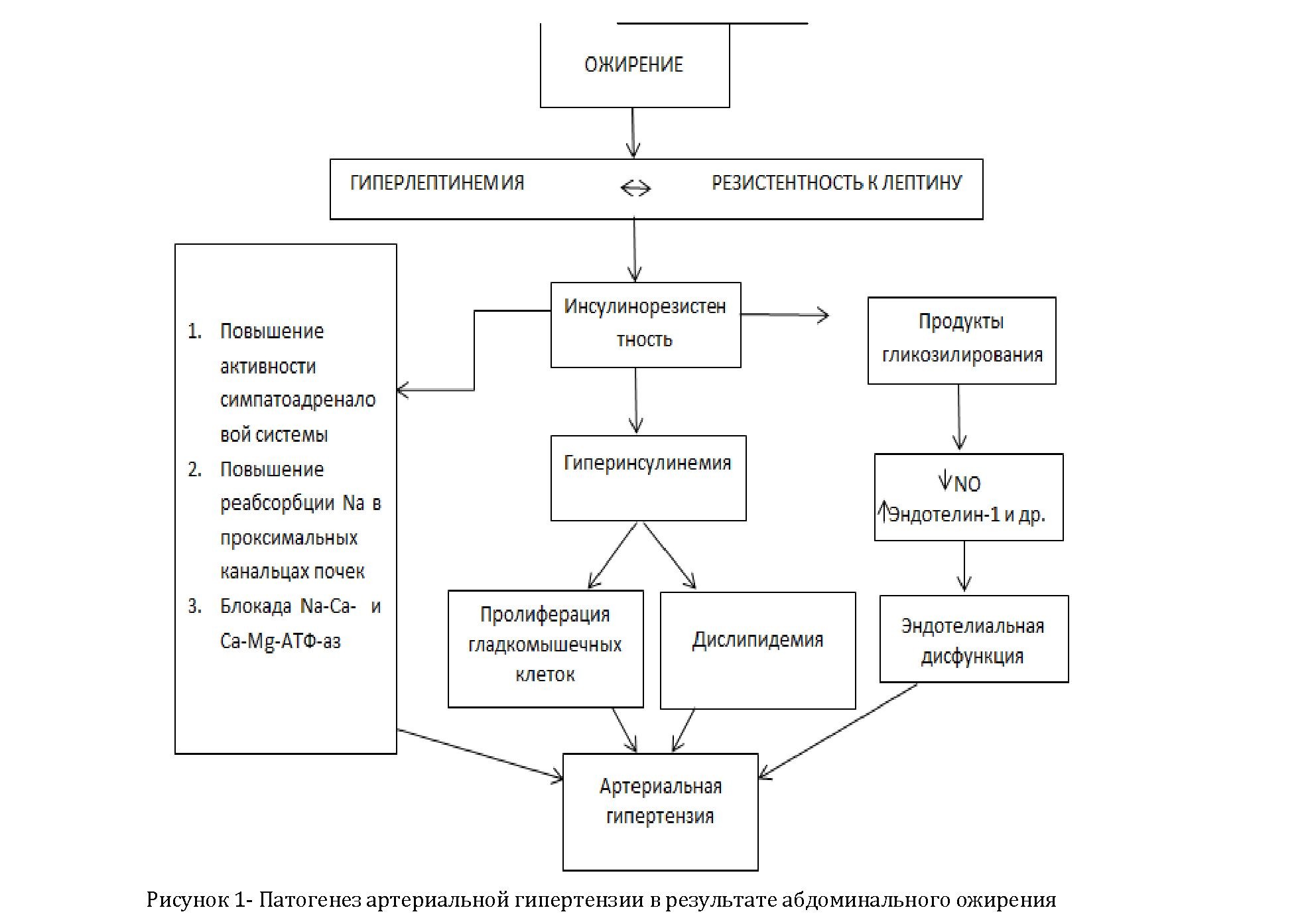
Артериальная гипертензия. Суммируя все вышеперечисленные факторы, становится понятен факт АГ у пациентов с МС. В следующей схеме изображены компоненты МС, которые приводят к развитию АГ. Патогенез артериальной гипертензии в результате абдоминального ожирения представлен на рисунке- 1.1.На рисунке 1 изображены основные компоненты, приводящие к АГ при МС. Однако вещества, продуцируемые адипоцитами, также влияют на развитие АГ при МС.
Оценка роли и функции жировой ткани как эндокринного органа очень важна в понимании патофизиологической взаимосвязи между ожирением и гипертонией. Почти все системные артерии окружены слоем периваскулярных жировой ткани (ПВЖТ). В большинстве миографических исследований, ПВЖТ удаляют, чтобы может предотвратить диффузию вазоактивных веществ. Из-за этого, о роли и функции ПВЖТ мало известно сосудистой биологии. ПВЖТ имеет модулирующее действия на сосудистую сократительную функцию. Это было описано в исследовании Soltis и Кассис, опубликованном в 1991 году[26]. Эти исследователи описали уменьшение чувствительности сосуда к норадреналину, когда аортальный сегменты остаются с ПВЖТ. Авторы демонстрируют, что это происходит из-за поглощения и элиминации катехоламинов жировой тканью. Но они не предположил, что ПВЖТ синтезирует какие-либо факторы [26].
Позднее в работах M. Löhn, было описано множество биоактивых веществ синтезируемых ПЖВХ [27,28,29,30,31,32]. Ниже будут описаны те, которые обладают определенными вазоактивных действия, а именно, лептин, адипонектин, TNF-α, простагландины, ангиотензин II и эндотелин-1.
 Лептин. Были обнаружены рецепторы к лептину в эндотелии сосудов. Это позволило найти взаимосвязь между ожирением и эндотелиальной дисфункцией [33]. Лептин является NO-зависимым вазодилататором при воздействии на эндотелиальные рецепторы, в то время как при воздействии на рецепторы расположенные в ЦНС он увеличивает периферическое сосудистое сопротивление и активность симпатической нервной [34]. Концентрация лептина в плазме коррелирует с ожирением, и гиперлептинемия считается независимой фактором риска ССЗ [35]. Выдвинуто 2 теории относительно вазорегуляции. Одно из них предполагает, что лептин участвует в регуляции сосудистого тонуса, одновременно вызывая нейрогенное прессорное действие и противоположный депрессорный эффект, опосредованный NO[36]. Согласно второй теории предполагается, что, лептин вызывает NO- зависимую вазодилатацию и, в то же время, его избыточное присутствие снижает эндотелий-зависимую релаксацию и приводит к эндотелиальной дисфункции. Нюансом второй теории заключается в том, что лептин-индуцированная релаксация происходит при концентрациях значительно выше тех, найденых у очень тучных субъектов [37]. Физиологические или патофизиологические концентрации лептина имеют лишь косвенное влияние на сосудистый тонус.
Лептин. Были обнаружены рецепторы к лептину в эндотелии сосудов. Это позволило найти взаимосвязь между ожирением и эндотелиальной дисфункцией [33]. Лептин является NO-зависимым вазодилататором при воздействии на эндотелиальные рецепторы, в то время как при воздействии на рецепторы расположенные в ЦНС он увеличивает периферическое сосудистое сопротивление и активность симпатической нервной [34]. Концентрация лептина в плазме коррелирует с ожирением, и гиперлептинемия считается независимой фактором риска ССЗ [35]. Выдвинуто 2 теории относительно вазорегуляции. Одно из них предполагает, что лептин участвует в регуляции сосудистого тонуса, одновременно вызывая нейрогенное прессорное действие и противоположный депрессорный эффект, опосредованный NO[36]. Согласно второй теории предполагается, что, лептин вызывает NO- зависимую вазодилатацию и, в то же время, его избыточное присутствие снижает эндотелий-зависимую релаксацию и приводит к эндотелиальной дисфункции. Нюансом второй теории заключается в том, что лептин-индуцированная релаксация происходит при концентрациях значительно выше тех, найденых у очень тучных субъектов [37]. Физиологические или патофизиологические концентрации лептина имеют лишь косвенное влияние на сосудистый тонус.
Адипонектин. Адипонектин - это секреторный белок, продуцируемый в больших количествах адипоцитов, который стабильно присутствует в плазме. У здоровых людей, адипонектин, предотвращает развитие сосудистых изменений, связаных с нарушениями липидного обмена, метаболизма глюкозы и резистентности к инсулину [38,39,40].
Концентрации адипонектина плазмы отрицательно коррелирует с индексом массы тела [41]. Было высказано предположение, что снижение концентрации адипонектина в плазме способствует развитию ИР и гиперинсулинемии. метаболических осложнений, связанных с ожирением. Таким образом, низкая концентрация адипонектина может быть предиктором метаболических нарушений и сосудистых изменений и, возможна, в фоновом режиме при АГ. Некоторые исследования показывают, что пациенты с АГ имеют более низкие концентрации адипонектина в плазме[42,43].
Фактор некроза опухоли^ (TNF-α). Исследователи команды Uysal K. показали, что гипертрофированные адипоциты синтезируют TNF-α . TNF-α- это один из многих факторов, вызывающих резистентность к инсулину. Поскольку ИР- это ключевой момент в развитии АГ у пациентов с МС, нельзя исключать роль TNF-α в ее патогенезе [44].
Простагландины, синтезированные адипоцитами. Простагландины являются наиболее вазоактивные веществами, генерируемые адипоцитов. Адипоциты синтезируют простагландины в ответ на симпатическую стимуляцию через ß-адренорецепторы. Этот стимул вызывает липолиз, высвобождение жирных кислот и простагландинов, особенно PGE2 и PGI 2. Адипоциты предоставляют арахидоновую кислоту для синтеза простагландинов, а необходимую циклооксигеназу они получают из соседних эндотелиальных клеток, т.к. циклооксигеназа в адипоцитах представленаменьших количествах, таких как простациклин (PGI 2)[45,46]. На том основании, что инсулин снижает производство этих мощных вазодилататоров, Паркер и его коллеги предположили, что АГ, связаная с ИР будет отчасти вызвана отсутствием надлежащего синтеза PGI2 [45]. Таким образом ИР провоцирует снижение синтез PGI2 и еще больше провоцирует развитие АГ.
Эндотелин-1, синтезируемый адипоцитами. Эндотелин-1 является сосудосуживающим белком, который синтезируется эндотелиальными клетками [47]. Существует также эндотелин-1 синтезируемый адипоцитами. В исследованиях T. Morise показано, что уровень эндотелина-1 увеличивается у пациентов с ожирением и диабетом 2 типа [48,49]. Harmelen и др. обнаружили, что пациенты с ожирением высвобождают из жировой ткани в 2,5 раза больше эндотелина-1, чем худые люди . Кроме того, эндотелин-1 участвует в развитии ИР [50].
Эти факты свидетельствуют в пользу положительной взаимосвязи ИР, МС и АГ.
Ангиотензин II, синтезируемый адипоцитами. Ангиотензин II- это сильнейший вазоконстриктор. Было предположение, что у тучных пациентов гиперактивирована РААС система, что приводит в высокой активности ренина плазмы и запускается производство ангиотензина II [51]. Но помимо этого есть еще 3 объяснения повышенных концентраций ангиотензина II:
- ожирение увеличивает секрецию ренина увеличением реабсорбции натрия в петле Генле [52];
- ожирение стимулирует секрецию ренина активацией симпатической нервной системы [53];
- наличие высокой активностью ренина в гипертрофированных адипоцитах вызывает повышенный синтез ангиотензина II [54].
Известно, что адипоциты синтезируют ангиотензин II [55]. Важно отметить, что экспрессия гена ангиотензиногена выше у внутрибрюшного жира, чем в других жировых депо или неадипозных тканей [56].
С метаболизмом ангиотензина II тесно связана концентрация альдостерона. Концентрации альдостерона повышены у некоторых тучных гипертоников, особенно у больных с висцеральным ожирением. Кроме того адипоциты самостоятельно синтезируют альдостерона в ответ на ангиотензин II [57,58].
Таким образом, МС со всеми своими основными компонентами и косвенными аспектами является независимым фактором риска развития ССЗ, в том числе и ишемического инсульта. Более того существуют исследования демонстрирующие МС как фактор бессимптомного поражения ГМ.
В 2008 году японские исследователи Hirokazu Bokura и соавторы опубликовали прорывную работу «Metabolic Syndrome Is Associated With Silent Ischemic Brain Lesions», в которой исследовали ассоциацию МС и 3 типа субклинического ишемического повреждения головного мозга (ГМ): бессимптомный инфаркт ГМ, перивентрикулярная гиперинтенсивность (ПГИ), и подкорковое повреждение белого вещества (ППБВ) [59].
Hirokazu Bokura и соавторы обследовали 1151 здоровых японца с МС при помощи МРТ и обнаружили три типа бессимптомных поражения. МС был диагностирован с использованием критериев АТР III [59].
После коррекции результата на возраст и другие факторы, МС оказался связан бессимптомным инфарктом ГМ, перивентрикулярной гиперинтенсивностью (ПГИ), и подкорковым повреждением белого вещества (ППБВ) с уровнем значимости 0,0001 [59].
Компоненты МС, такие как повышенное АД коррелировало со всеми видами поражений, а дислипидемии и повышенный уровень глюкозы натощак были ассоциированы с подкорковых повреждения белого вещества и перивентрикулярной гиперинтенсивностью, соответственно. Наличие и распространенность бессимптомных поражений ГМ зависело от количества компонентов МС. В заключение авторы заявили, что компоненты МС и бессимптомные повреждения могут быть использованы в качестве диагностических инструментов для прогнозирования и предотвращения развития будущего инсульта [59].
До Hirokazu Bokura было опубликовано всего 2 работы посвященные изучению взаимосвязи между МС и бессимптомным поражением головного мозга.
В 2014 году Michiel Sala и соавторы в работе «Microstructural Brain Tissue Damage in Metabolic Syndrome» исследовали связь между компонентами метаболического синдрома и целостностью ткани ГМ с помощью магнитно-резонансной томографии. Обследованию подверглись участники Leiden Longevity Study. Было включено 130 субъектов. Метаболический синдром оценивался критериями АТР III. Был проведен линейный и логистический регрессионный анализ, чтобы оценить связь между МС и макроструктурой ГМ (объем мозга, поражение белого вещества, лакунарные инфаркты, мозговые микрокровоизлияния) и микроструктура (средний коэффициент передачи намагничивания, гистограмма высот пика, дробная анизотропия, и средняя диффузия). Также применили линейный и порядковый регрессионный анализ, чтобы определить индивидуальный вклад одного компонента МС в развитии четырех других параметров. Модели были скорректированы по возрасту и полу. Макроструктура мозга не была связана с МС. Но компоненты МС холестерина ЛПВП, триглицериды, ИМТ и диастолическое артериальное давление были независимыми факторами изменений в мозговой микроструктуре. Авторы пришли к выводу, что МС, является независимым фактором риска целостности тканей мозга. А при раннем развитии МС может быть обнаружена утрата мозговой ткани [60].
Заключение. Таким образом, знание этиопатогенетических принципов развития ишемического инсульта является важным аспектом для врача любого профиля в связи с широким распространением факторов риска среди населения. Пациентам с метаболическим синдромом необходимо проводить лечение, направленное на снижение веса, АД, нормализацию уровня глюкозы и холестерина с целью профилактики сердечно-сосудистых заболеваний.
СПИСОК ЛИТЕРАТУРЫ
- Кадырова И.А. Разработка математической модели вероятности развития острого нарушения мозгового кровообращения у пациентов с метаболическим синдромом: дис. ... д-ра мед.наук - Караганда, 2016. - 72 с.
- Kylin E. Zur Frage der Adrenalinreaktion. Zeitschrift für Die Gesamte // Experimentelle Medizin. - 1925. - №44(1).- Р. 227-239.
- Vague J. Presse Medl.- 1947. - 340 р.
- Reaven G. Role of Insulin Resistance in Human Disease // Diabetes. - 1988. - №37(12) .- Р1595-1607.
- Kaplan N. The Deadly Quartet //Arch Intern Med.-1989.- №149(7) .-Р.1514-1518.
- World Health Organization. Definition, Diagnosis and Classification of Diabetes Mellitus and its Complications. - 1999. - 88 р.
- Balkau B. Diabetes epidemiology: present and future // Diabetic Medicine.- 1999. -№16(6) .- Р.446-447.
- Carlos Lorenzo, Ken Williams, Kelly J. Hunt. Executive Summary of the Third Report of The National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol In Adults (Adult Treatment Panel III) // JAMA.- 2001.-№ 285.- Р. 2486-2497.
- P. Poirier, T.D. Giles, G.A. Bray, et al. Obesity and cardiovascular disease: pathophysiology, evaluation, and effect of weight loss: an update of the 1997 American Heart Association scientific statement on obesity and heart disease from the obesity committee of the council on nutrition, physical activity, and metabolism // Circulation.- 2006.-№ 113.- Р. 898-918.
- P.J. Enriori, A.E. Evans, P. Sinnayah, M.A. Crowley Leptin resistance and obesity // Obesity.- 2006.- № 14 .- Р. 254-258.
- A. Romero-Corral, J. Sierra-Johnson, F. Lopez-Jimenez, et al. Relationships between leptin and C-reactive protein with cardiovascular disease in the adult general population // Nat Clin Pract Cardiovasc Med.- 2008.- №5.- Р. 418-425.
- C.J. Lavie, R.V. Milani, H.O. Ventura. Untangling the heavy cardiovascular burden of obesity // Nat Clin Pract Cardiovasc Med.- 2008.- № 5.- Р. 428-429.
- Alpert M.A. Obesity cardiomyopathy: pathophysiology and evolution of the clinical syndrome //Am. J. Med. Sc.i.- 2001.- № 321.- Р. 225236.
- Messerli F.H., B.D. Nunez B.D., Ventura H.O. Overweight and sudden death: increased ventricular ectopy in cardiomyopathy of obesity // Arch Intern Med.- 1987.-№ 147.-Р. 1725-1728.
- C.J. Lavie, R.V. Milani, H.O. Ventura, G.A. Cardenas, M.R. Mehra, F.H. Messerli. Disparate effects of left ventricular geometry and obesity on mortality in patients with preserved left ventricular ejection fraction // Am J Cardiol.-2007.-№ 100.- Р. 1460-1464.
- Lavie C, Milani R, Ventura H, Cardenas G, Mehra M, Messerli F. Disparate Effects of Left Ventricular Geometry and Obesity on Mortality in Patients With Preserved Left Ventricular Ejection Fraction // The American Journal of Cardiology.- 2007.- №100(9) .- Р.1460-1464.
- T. Kurth T., Gaziano J.M., Berger K., et al. Body mass index and the risk of stroke in men //Arch Intern Med.- 2002.- №162.- Р. 2557-2562. Дедов И.И., Шестакова М.В. Сахарный диабет. - М.: Универсум Паблишинг, 2003. - 222 с.
- МакДермотт М.Т. Секреты эндокринологии.- М.: Бином, 2001. - 39 с.
- Титов В.Н., Хохлова Н.В., Ширяева Ю.К., Глюкоза, гликотоксины и продукты гликирования протеинов: роль в патогенезе // Клиническая медицина.- 2013.- № 3.- С.15-24.
- Мкртумян А.М., Полукаров М., Стрюк Р.И., Давыдов А.Л. Диабетическая макроангиопатия. Возможность доклинической диагностики // Болезни сердца и сосудов.-2008.- №2.-С. 16-20.
- Evans M., Anderson RA., Graham J et al. Ciprofi brate therapy improves endothelial function and reduces postprandial lipemia and oxidative stress in type 2 diabetes mellitus //Circulation .- 2000.-№101(15) .- Р.1773-9.
- Оганов Р.Г, Масленникова Г.Я. Смертность от сердечно-сосудистых и других хронических неинфекционных заболеваний среди трудоспособного населения России// Кардиоваскул. тер.и профилак.- 2002.- № 3.-С. 4-8.
- Рунихин А.Ю. Синдром гипергликемии в практике кардиолога // Кардиология.- 2005.-№ 10.-С.85-90.
- Северина А. С., Шестакова М. В. Нарушение системы гемостаза у больных сахарным диабетом // Сахарный диабет.- 2004.- №1.- С.62-67.
- Soltis E. E., Cassis L. A., Influence of perivascular adipose tissue on rat aortic smooth muscle responsiveness // Clinical and Experimental Hypertension .- 1991. - Vol. 13, № 2.- Р. 277-296.
- Löhn M., Dubrovska G., Lauterbach B., Luft F. C. Periadventitial fat releases a vascular relaxing factor // The FASEB Journal.- 2002.- Vol. 16, № 9.-Р. 1057-1063.
- Verlohren S., Dubrovska G., Tsang S. Y. et al.Visceral periadventitial adipose tissue regulates arterial tone of mesenteric arteries // Hypertension.- 2004.- Vol. 44, № 3.-Р. 271-276.
- Maenhaut N., J. Van deVoorde. Regulation of vascular tone by adipocytes// BMC Medicine.- 2011.- №9. - Р. 25-31.
- Gao Y. J., Lu C., Su L. Y. Modulation of vascular function by perivascular adipose tissue: the role of endothelium and hydrogen peroxide // British Journal of Pharmacology. - 2007. - №3.- Р. 323-331 .
- Gao Y. J., Zeng Z. H., Teoh K. et al. Perivascular adipose tissue modulates vascular function in the human internal thoracic artery // Journal of Thoracic and Cardiovascular Surgery.- 2005.- № 4.- Р. 1130-1136.
- Schleifenbaum J., Köhn, N. Voblova et al. Systemic peripheral artery relaxation by KCNQ channel openers and hydrogen sulfide // Journal of Hypertension.- 2010.- № 9.- Р. 1875-1882.
- Winters B., Mo Z., Brooks E. Reduction of obesity, as induced by leptin, reverses endothelial dysfunction in obese (Lep(ob)) mice // Journal of Applied Physiology.- 2000.- №6.- Р. 2382-2390.
- Shirasaka T., Takasaki M., Kannan H. Cardiovascular effects of leptin and orexins // American Journal of Physiology.- 2003.-№ 3.- Р. 639651.
- Considine R. V., Sinha M. K., Heiman M. L. et al. Serum immunoreactive-leptin concentrations in normal-weight and obese humans // The New England Journal of Medicine.- 1996.- № 5.- Р. 292-295.
- Frühbeck G. Pivotal role of nitric oxide in the control of blood pressure after leptin administration // Diabetes.-1999.- № 4.- Р. 903-908. Knudson J. D., Dincer U. D., Zhang C. et al. Leptin receptors are expressed in coronary arteries, and hyperleptinemia causes significant coronary endothelial dysfunction // American Journal of Physiology.-2005.- № 1.- Р. 48-56.
- Yamauchi T., Kamon J., Waki H. et al. Тhe fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity// Nature Medicine.- 2001.-№ 8.- Р. 941-946.
- Yamauchi T., Kamon J., Minokoshi Y. et al. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase // Nature Medicine.- 2002.- №. 11.- Р. 1288-1295.
- Kondo H., Shimomura L., Matsukawa Y. et al. Association of adiponectin mutation with type 2 diabetes: a candidate gene for the insulin resistance syndrome // Diabetes.-2002 .- № 7.- Р. 2325-2328.
- Takahashi M., Funahashi T., Shimomura I. Plasma leptin levels and body fat distribution // Hormone and Metabolic Research.- 1996.- № 12.- Р. 751-752 .
- Asayama K., Hayashibe H., Dobashi K. et al. Decrease in serum adiponectin level due to obesity and visceral fat accumulation in children // Obesity Research.- 2003.- № 9.- Р. 1072-1079.
- Mallamaci F., Zoccali C., Cuzzola F. et al. Adiponectin in essential hypertension // Journal of Nephrology.- 2002.- № 5.- Р. 507-511.
- Uysal K. T., Wiesbrock S. M., Marino M. W. Рrotection from obesity-induced insulin resistance in mice lacking TNF- α function // Nature.- 1997.- № 6651.- Р. 610-614.
- Parker J., Lane J., Axelrod L. Cooperation of adipocytes and endothelial cells required for catecholamine stimulation of PGI2 production by rat adipose tissue// Diabetes.- 1989.- № 9.- Р. 1123-1132.
- Richelsen B., Borglum J. D., Sorensen S. S. Biosynthetic capacity and regulatory aspects of prostaglandin E2 formation in adipocytes // Molecular and Cellular Endocrinology.- 1992.- № 1-2.-Р. 73-81.
- Fain J. N., Tagele B. M., Cheema P. Release of 12 adipokines by adipose tissue, nonfat cells, and fat cells from obese women// Obesity.- 2010.- № 5.-Р. 890-896.
- Morise T., Takeuchi Y., Kawano M. Increased plasma levels of immunoreactive endothelin and von Willebrand factor in NIDDM patients // Diabetes Care.- 1995.- № 1.- Р. 87-89.
- Da Silva A., Kuo J. J., Tallam L. S., Hall J. E. Role of endothelin-1 in blood pressure regulation in a rat model of visceral obesity and hypertension // Hypertension.- 2004.- № 2.- Р. 383-387.
- Van Harmelen V., Eriksson A., Åström G. et al.Vascular peptide endothelin-1 links fat accumulation with alterations of visceral adipocyte lipolysis // Diabetes.-2008.- № 2.- Р. 378-386 .
- Tuck M. L., Sowers J., Dornfeld L. The effect of weight reduction on blood pressure, plasma renin activity, and plasma aldosterone levels in obese patients // The New England Journal of Medicine.- 1981.- № 16.- Р. 930-933.
- Hall J. E. Mechanisms of abnormal renal sodium handling in obesity hypertension //American Journal of Hypertension.-1997.- № 5.- Р. 49-55.
- Hall J, Brands M, Hildebrandt D, Mizelle H. Obesity-associated hypertension. Hyperinsulinemia and renal mechanisms // Hypertension.- 1992.- №19.- Р.145-152.
- Engeli S., Schling P., Gorzelniak K. et al., The adipose-tissue renin-angiotensin-aldosterone system: role in the metabolic syndrome?// International Journal of Biochemistry and Cell Biology.-2003.- № 6.- Р. 807-825.
- G|lvez-Prieto B., Bolbrinker J., Stucchi P. et al. Comparative expression analysis of the renin—angiotensin system components between white and brown perivascular adipose tissue // Journal of Endocrinology.- 2008.- № 1.- Р. 55-64.
- Rahmouni K., Mark A. L., Haynes W. G., Sigmund C. Adipose depot-specific modulation of angiotensinogen gene expression in diet- induced obesity // American Journal of Physiology.- 2004.- № 6.- Р. 891-895.
- Goodfriend T. L., Calhoun D. A. Resistant hypertension, obesity, sleep apnea, and aldosterone: theory and therapy // Hypertension.- 2004.- № 3.- Р. 518-524.
- Briones A. M., Nguyen Dinh Cat A., Callera G. E., et al. Adipocytes produce aldosterone through calcineurin-dependent signaling pathways //Hypertension.-2012.- № 5.- Р. 1069-1078.
- Bokura H, Yamaguchi S, Iijima K., Nagai A, Oguro H. Metabolic Syndrome Is Associated With Silent Ischemic Brain Lesions // Stroke.- 2008.- № 39(5) .- Р. 1607-1609.
- Sala M, de Roos A, van den Berg A et al. Microstructural Brain Tissue Damage in Metabolic Syndrome // Diabetes Care. - 2013. - № 37(2). - Р.493-500.