Мукополисахаридозы (МПС) –наследственные болезни накопления, обусловленные нарушением обмена ГАГ, по причине генетическойнеполноценности лизосомальных ферментов, участвующих в их расщеплении. При МПС нерасщепленные ГАГ активно аккумулируют в соединительной ткани организма.Накопленные ГАГ запускают аутоиммунный воспалительный процесс путем активации Толл подобных рецепторов 4, стимулирования воспалительных цитокинов (ФНО-a, ИЛ-1ß) и активации Деструктивных энзимов (MMP 13, катепсин СТ) и NO, что является причиной развития апоптозахондроцитов. Результатом апоптоза, индуцированного накоплением ГАГ является деструкция и низкая регенерация костно-суставной ткани.
Актуальность: Мукополисахаридозы (МПС) – болезни накопления, обусловленные нарушением обмена гликозаминогликанов (ГАГ) в результате генетической неполноценности лизосомальных ферментов, участвующих в их расщеплении [1]. МПС относятся к редким, орфанным заболеваниям, распространенность варьирует от типа МПС и составляет 1 случай на 20000 – 100000 новорожденных. Наблюдаемая при МПС неполноценность названных ферментов, приводит к нарушению деградации и активному накоплению ГАГ в лизосомах соединительной ткани. В связи с чем, при всех типах МПС костно-суставная система вовлекается в патологический процесс, имеющий прогрессирующий характер, вплоть до ее деструкции [2]. Поражение костно-суставной системы является причиной развития ранней инвалидизации, в связи, с чем необходимо изучить патогенез повреждения костно-суставной системы при МПС с целью разработки патогенетической терапии.
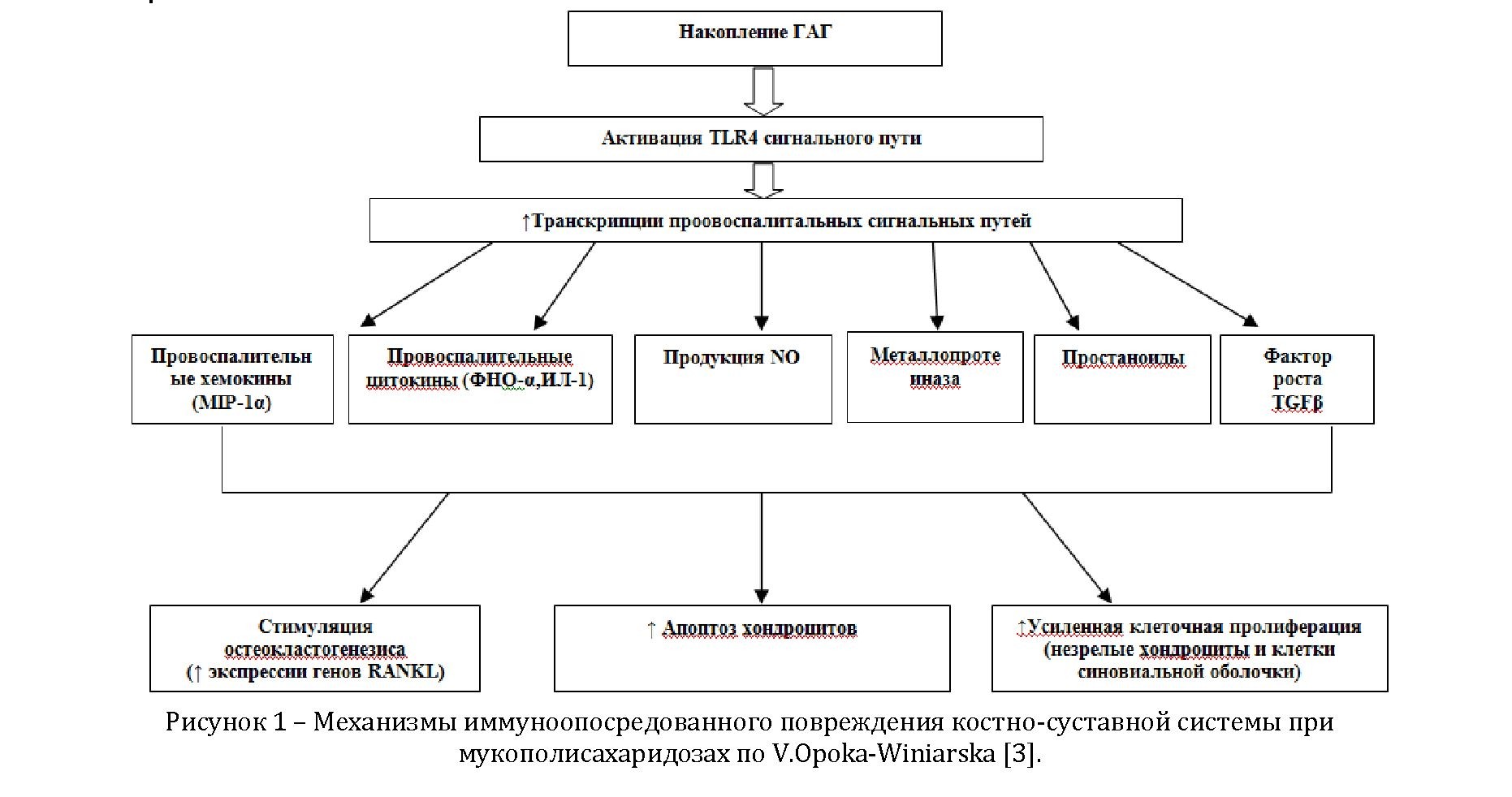
При МПС идет активное накопление нерасщепленных ГАГ во всех соединительнотканных структурах. В зависимости от типа накапливающегося ГАГ (дерматан сульфат, кератан сульфат, хондроитин сульфат, гепаран сульфат) в настоящее время различают 7 типов МПС: I, II, III, IV, VI, VII и IX. ГАГ - полисахариды, представляющие собой углеводную часть протеогликанов. Продукция ГАГ в виде полисахаридных цепей происходит в аппарате Гольджи фибробластов. ГАГ, являясь гетерополисахаридами, принимают участие в организации внутриклеточного матрикса, как основное вещество, и обеспечивают межклеточные коммуникации. Важной частью обмена ГАГ, является их распад, который происходит в лизосомах под воздействием таких ферментов как эндогликозидаза, экзогликозидаза и сульфатаза. ГАГ, взаимодействуя с большим количеством протеинов (включая хемокины, цитокины, факторы роста, морфогены, ферменты и адгезивные молекулы) индуцируют воспаление в разных клетках [3]. Так, накопленные ГАГ, связываясь с Толл подобными рецепторами 4 (TLR4), запускают воспалительные процессы путем стимулирования воспалительных цитокинов, таких как ФНО-a, ИЛ-1ß и NFkB, активирования деструктивных энзимов MMP 13, катепсин СТ и NO, что приводят к апоптозу хондроцитов. Апоптоз, индуцированный накоплением ГАГ, является ведущим фактором в деструкции хрящевой ткани и дегенеративном изменении суставов при МПС [3]. M.Simonaro доказано, что в патогенезе МПС основным путем воспаления является TLR4 опосредованный путь. В норме активация TLR4 сигнального пути индуцируется воздействием липополисахаридов (ЛПС), являющихся основным компонентом клеточной стенки грамотрицательных бактерий. ЛПС увеличивают экспрессию генов кодирующих TLR4, посредством связывания и активации генов LBP (ЛПС связывающий белок), CD14 (мембранный гликозилфосфатидилинозитол- связанный белок, компонент рецепторного комплекса CD14/TLR4/MD2, распознающего ЛПС) и MyD88 (myeloid differentiation primary response gene 88, цитозольный адаптерный белок), участвующих в передаче сигнала от TLR [4]. Cтруктурная схожесть эндогенных фрагментов ГАГ, в частности дерматан сульфата, с ЛПС, приводит при МПС к активации TLR4 сигнального пути. Причем доказано, что дерматан сульфат стимулирует высвобождение NO из здоровых клеток в большей степени, чем ЛПС [5]. В исследованиях M.Simonaro, проведенных на крысах, было продемонстрировано, что дерматан сульфат так же как ЛПС может стимулировать высвобождение NO и, тем самым, вызывать апоптоз хондроцитов [4].
Результатом активации TLR4 пути является синтез и секреция воспалительных медиаторов, включая хемокины (MIP1α), цитокины (ФНО-a и ИЛ1) и матриксную металлопротеиназу (MMP). В свою очередь, ГАГ усиливают экспрессию генов CD44 и MyD88, что является достаточным для активации TLR4 сигнала. В экспериментальных исследованиях при инактивации TLR4 у мышей с МПС VII типа была отмечена положительная динамика в отношении роста и плотности костных пластинок и, самое главное, происходила нормализация уровня ФНО-a [5].
В исследованиях Wang было выявлено, что введение здоровым мышам ГАГ, таких как гиалуроновая кислота, гепаран сульфат, кератан сульфат, индуцирует у них клинику воспаления в виде артрита, тендосиновиита, дерматита и клеточную инфильтрацию в различные соединительнотканные структуры. Циркулирующие или локально освобожденные ГАГ стимулируют клональную экспансию различных клеток, включая Т-лимфоциты, В- лимфоциты и макрофаги (МФ). Эти данные указывает на запуск процессов иммунизации против собственных антигенов, таких как ГАГ, что изменяет в организме иммунный ответ и вызывает хронический воспалительный процесс [6]. Так как избыток ГАГ при МПС является метаболическим фактором запуска воспалительного процесса, G.S.Hotamisligil предложил термин metaflammation - воспаление запущенное метаболическим процессом [7].
Di Rosario в своих работах по изучению головного мозга мышей с МПС III-В типа продемонстрировал повышенное стимулирование множественных иммуно-связанных генов врожденного и адаптивного иммунитета, включая гены Т- лимфоцитов, В-лимфоцитов, комплемент, иммуноглобулины, TLR и антиген презентирующие молекулы [8]. Наряду с этим, S.Killedar установил, что введение лимфоцитов мышей с МПС III В типа здоровым мышам вызывает у последних нейровоспаление с дальнейшим развитием клиники поражения центральной нервной системы. Также им было выявлено значительное увеличение экспрессии генов цитокинов: IFN-γ, IFN-α, ИЛ-2, ИЛ-4, ИЛ-5, ИЛ-17 и ФНО-a. Отмечалось увеличение в нейроглие CD3e, CD4 и CD8a, что свидетельствует об инфильтрации Т-лимфоцитами клеток головного и спинного мозга мышей с МПС III. Эти данные позволяют предположить, что накапливающиеся гепаран сульфат при МПС III инициирует патологический иммунный ответ и запускает аутоиммунный процесс, который в дальнейшем приводит к нейровоспалению [9].
При МПС синовиальная мембрана гиперплазирована, что обусловлено более быстрой (по сравнению со здоровыми) пролиферацией синовиальных клеток, как результат повышенных уровней ФНО-a, ИЛ-1 и TGFß. В свою очередь TGFβ представляет из себя белок, который контролирует пролиферацию и клеточную дифференцировку незрелых хондроцитов и синовиоцитов. В исследованиях M.Simonaro установлено, что, несмотря на активацию провоспалительных цитокинов, приводящих к апоптозу хондроцитов, в целом не отмечается значительного снижения количества последних, что объясняется увеличенной экспрессией TGFβ. Однако, низкий уровень остеонектина, как следствие остеопении, приводит к недостаточному связыванию кальция в костях и уменьшению их минерализации, синтезу незрелых хондроцитов, не способных сформировать здоровую костную ткань [10]. В свою очередь, развитие остеопении связано с повышением уровня остеокластов, являющихся медиаторами воспаления и эрозии костной ткани. При МПС остеопения поддерживается большим количеством остеокластов и их предшественников в костном мозге. Остеокластогенезис зависит от взаимодействия RANK и его лиганды RANKL. Экспрессия генов RANKL регулируется цитокинами ФНО-a, ИЛ1-ß. Известно, что синовиоциты у больных МПС секретируют RANK, который привлекает макрофаги из крови и стимулирует их дифференцировку в остеокласты, которые приводят к остеопении.
RANK - рецептор активатор NFkB (ядерный фактор транскрипции kappa В), так же известен как TRANCE рецептор, является членом семейства ФНО-a рецепторов. RANK является рецептором для RANK лиганда (RANKL) и частью RANK/RANKL/OPG опосредованного пути, который регулирует дифференцировку и активацию остеокластов. OPG - это рецептор приманка для RANK, регулирует стимуляцию RANK сигнального пути, конкурируя за RANKL. Значительное увеличение экспрессии RANKL гена было установлено в хондроцитах, синовиацитах и фибробласт- подобных синовиацитах у крыс с МПС VI типа [11]. Провоспалительные цитокины, такие как ИЛ1ß и ФНО-a являются мощными стимуляторами резорбции костных остеокластов путем индукции экспрессии RANKL и активации рецепторов на поверхности моноцитов. Исследования на животных с МПС VI и VII показали, что при МПС предшественники остеокластов и другие клетки макрофагального происхождения могут высвобождать провоспалительные цитокины и поддерживать остеокластогенезис. Вышеупомянутый NF-κB - универсальный фактор транскрипции, контролирующий экспрессию генов иммунного ответа. апоптоза и клеточного цикла. Нарушение регуляции NF-kB вызывает воспаление и аутоиммунные заболевания. NF-kB активируется цитокинами, такими как ФНО-a, ИЛ1 [12].
Медиаторы воспаления, активированные TLR4- опосредованным путем, оказывают большое влияние на увеличение апоптоза хондроцитов и изменения в матриксе путем стимулирования экспрессии генов на матриксной металлопротеиназе (MMP), что было доказано на животных с МПС VI и VII типами. MMP относится к семейству внеклеточных цинк-зависимых эндопептидаз, способных разрушать все типы белков внеклеточного матрикса, вызывая тем самым апоптоз. MMP является первично ответственным за деградацию хрящевого матрикса, к нему относятся MMP 1, 8 и 13. Немаловажную роль в развитие артрита играет катепсин СТ, который существенно экспрессируется и увеличивается в течение воспалительного процесса в хондроцитах, остекластах, синовиальных фибробластах, макрофагах и лейкоцитах. При МПС накопление деструктивных энзимов, таких как катепсин СТ и MMP, в синовиальных фибробластах и остеокластах значительно усиливается под воздействием воспалительных цитокинов ФНО-a и ИЛ1, что приводит к разрушению ключевых компонентов экстрацелюлярного матрикса, включая эластан, коллагены и протеогликаны. [4]. В настоящее время существует также теория регуляции экспрессии TLR4 путем инактивации JAK-STAT механизма. Путь JAK-STAT является основным путем внутриклеточной передачи сигналов с рецепторов цитокинов. Существует две основные молекулы этого пути - киназа JAK и транскрипционный фактор Signal Transducer and Activator of Transcription (STAT). Существует несколько типов STAT: STAT1. STAT2. STAT3. STAT4. STAT5. STAT6 и STAT7. В механизме развития воспалительного процесса при МПС задействованы 2 типа STAT. Анти-пролиферативный эффект на рост пластинок хондроцитов оказывает STAT1, в то время как STAT3 является про-пролиферативным. В работах
J.A.Metcalf отмечалось снижение пролиферации хондроцитов и количества растущих пластинок при МПС VII типа, ассоциированных со снижением экспрессии STAT3. Вероятно, уменьшение активности STAT3 является механизмом, приводящим к снижению пролиферации клеток костно-хрящевой системы [13].
Выводы: таким образом, накопление ГАГ запускает каскад взаимосвязанных метаболических, воспалительных и иммунологических ответов с системным эффектом, приводящий к аутоиммунному воспалению и апоптозу хондроцитов, что, в конечном итоге, приводит к низкому качеству регенерации костно-хрящевой ткани (рисунок 1). Следует отметить, что все вышеуказанные данные, являются результатами экспериментальных исследований на животных. Аналогичные исследования с участием людей не проводились. Вместе с тем, результаты проведенных исследований являются доказательством того, что при МПС имеет место системное хроническое воспаление, схожее с воспалительным процессом при многих аутоиммунных заболеваниях (ревматоидный артрит, склеродерма, системная красная волчанка), связанное с патологическим метаболизмом ГАГ.
В настоящее время в качестве заместительной терапии МПС II. IV-A и VI типов используются рекомбинантные лизосомальные ферменты. Однако данный вид терапии не дает регрессии уже развившегося поражения костносуставной системы. Выявленные в экспериментальных исследованиях особенности иммунопатогенеза при МПС дают основание для разработки новых методов терапии данной патологии - с применением противовоспалительных препаратов, ингибирующих ФНО-a - основного воспалительного цитокина [9].
СПИСОК ЛИТЕРАТУРЫ
- Atul Mehta, Bryan Winchester. Lysosomal storage disorders.//A practical guide. Wiley-Blackwell. - 2012. - C. 94-100.
- Lorne A.Clarke. Pathogenesis of skeletal and connective tissue involvement in the mucopolysaccharidoses: gycosaminoglycan storage is merely the instigator.//Rheumatology Journal. - 2011.- №50. -C.13-18.
- V.Opoka-Winiarska, A.Jurecka, A.Emeryk, A.Tylki-Szymanska. Osteoimmunology in mucopolysaccharidosis type I, II, VI and VIII. Immunological regulation of the isteoarticular system in the course of metabolic inflammation.//Osteoarthritis and Cartilage Journal. - 2013.- №21.-С. 1813-1823.
- Simonaro CM, Ge Y. Eliyahu, He X.Jepsen, Schichman EH. Involvement of the Toll like receptor 4 pathway and use of TNF-α antagonists for treatment of the mucopolysaccharidosis.//Proc National Academy Science USA. - 2010. -№1. -107. -С. 222-227.
- Simonaro CM, Haskins ME, Schichman EH. Articular chondrocytes from animals with dermatan sulfat storage disease undergo a high rate of apoptosis and release nitric oxide and inflammatory cytokins: a possible mechanismunderlying degenerative joint disease in the mucopolysaccharidosis.//Laboratory Investigation Journal. - 2001. - №9. - С. 1319-1328.
- Wang JY, Roehrl MH. Glucosaminoglycans are potential cause of rheumatoid arthritis. Proc National Academy Science USA. - 2002. - №29. - C.14362-7.
- Hotamisligi GS. Inflammation and metabolic disorders.//Nature Journal. - 2006.- №444.-С. 860-867,
- DiRosario J, Divers E, Wang C, Etter J, Charrier A. Innate and adaptive immune activation in the brain of MPS IIIB mouse model.//Neuroscience Research Journal. - 2009. -№89. –С. 978-990.
- Killedar S, DiRosario J, Divers E, Popovich PG, McCarty DM, Fu H. Mucopolysaccharidosis IIIВ, a lysosomal storage diseases, trigger a pathogenetic СNS autoimmune response.//Journal of Neuroinflammation. - 2010. -№7. 39 с.
- Simonaro CM, D'Angelo M, Haskins ME, He X, Eliyahu E. Mechanism of glycosaminoglycan-mediated bone and joint disease: implication for mucopolysaccharidoses and other connective tissue diseases.,,American Journal of Pathology.- 2008. -№57. –С.112-121.
- Simonaro CM, D'Angelo M, Haskins ME, Schuchman E.H. Joint and bone disease in mucopolysaccharidoses VI and VII: identification of new therapeutic targets and biomarkers using animals models.//Pediatric Research Journal. - 2005. -№172. –С. 701-707.
- Monroy MA, Ross FP, Teitelbaum SL, Sands MS. Abnormal osteoclast morphology and bone remodeling in a murine model of lysosomal storage diseas.//Bone Journal. –2002.-№30. – С. 532-359.
- Metcalf JA, Zhang Y, Hilton MJ, Long F, Ponder KP. Mechanism of shortened bones in mucopolysaccharidosis type VII.//Molecular Genetic Metabolism. – 2009. - №97. –С.202-211.