Подходы к процедуре определения и оценке приемлемости прецизионности УФ-спектрофотометрических методик количественного определения аналитов в биологических жидкостях, применяемых в судебно-токсикологическом анализе, предложенные авторами в предыдущей работе, апробированы на примере УФ-спектрофотометрической методики количественного определения доксиламина в крови.
Введение. Данная статья является продолжением работы авторов [1 - 4, 6, 7] в области разработки подходов к валидации методик количественного определения для целей судебно-токсикологического анализа и посвящена вопросам разработки процедуры определения и формирования критериев приемлемости для валидационного параметра «прецизионность».
Целью данной работы является апробация подходов к процедуре определения и оценке приемлемости прецизионности УФ-спектрофотометрических методик количественного определения аналитов в биологических жидкостях, применяемых в судебно-токсикологическом анализе, предложенных в нашей предыдущей работе [4], на примере УФ-спектрофотометрической методики количественного определения доксиламина в крови.
Материалы и методы исследования. Рабочие растворы: 1000,0 мг доксиламина сукцината вносили в мерную колбу емкостью 250,0 мл, растворяли в воде очищенной и доводили объем раствора этим же растворителем до метки (стандартный раствор 1, концентрация 4000 мкг/мл). В семь мерных колб емкостью 100,0 мл вносили из бюретки 32,50; 30,00; 25,00; 20,00; 15,00; 10,00 и 5,00 мл стандартного раствора доксиламина сукцината 1 соответственно и доводили объемы растворов водой очищенной до метки (рабочие растворы 1, 2, 3, 4, 5, 6 и 7 соответственно, концентрация 1300, 1200, 1000, 800, 600, 400 и 200 мкг/мл соответственно).
400,0 мг доксиламина сукцината вносили в мерную колбу емкостью 100,0 мл, растворяли в воде очищенной и доводили объем раствора этим же растворителем до метки (стандартный раствор 2, концентрация 4000 мкг/мл). В пять мерных колб емкостью 100,0 мл вносили из бюретки 32,50; 30,00; 20,00; 10,00 и 5,00 мл стандартного раствора доксиламина сукцината 2 соответственно и доводили объемы растворов водой очищенной до метки (рабочие растворы 8, 9, 10, 11 и 12 соответственно, концентрация 1300, 1200, 800, 400 и 200 мкг/мл соответственно).
Модельные растворы: 100,0 мг доксиламина сукцината вносили в мерную колбу емкостью 500,0 мл, растворяли в 0,1 моль/л растворе кислоты хлористоводородной и доводили объем раствора этим же растворителем до метки (стандартный раствор 3, концентрация 200 мкг/мл). В семь мерных колб емкостью 100,0 мл вносили из бюретки 26,00; 24,00; 20,00; 16,00; 12,00; 8,00 и 4,00 мл стандартного раствора доксиламина сукцината 3 соответственно и доводили объемы растворов 0,1 моль/л раствором кислоты хлористоводородной до метки (модельные растворы 1, 2, 3, 4, 5, 6 и 7 соответственно, концентрация 52, 48, 40, 32, 24, 16 и 8 мкг/мл соответственно).
Раствор сравнения: 400,0 мг доксиламина сукцината вносили в мерную колбу емкостью 100,0 мл, растворяли в 0,1 моль/л растворе кислоты хлористоводородной и доводили объем раствора этим же растворителем до метки (стандартный раствор 4, концентрация 4000 мкг/мл). В мерную колбу емкостью 100,0 мл вносили из бюретки 18,00 мл стандартного раствора доксиламина сукцината 4 и доводили объем раствора 0,1 моль/л раствором кислоты хлористоводородной до метки (стандартный раствор 5, концентрация 720 мкг/мл). В мерную колбу емкостью 50,0 мл вносили 2,00 мл стандартного раствора доксиламина сукцината 5 и доводили объем раствора 0,1 моль/л раствором кислоты хлористоводородной до метки (раствор сравнения, концентрация 28,8 мкг/мл).
Калибровочные образцы: 3 серии по 7 образцов (20,00 мл) модельной крови (матрица), полученной от 3 различных источников, в которые введено по 1,00 мл рабочих растворов 1 - 7 соответственно.
Модельные образцы: 3 серии по 5 образцов (20,00 мл) модельной крови, полученной от 3 различных источников, в которые введено по 1,00 мл рабочих растворов 8-12 соответственно;
Анализируемые растворы: полученные по валидируемой методике [3] растворы для калибровочных и модельных образцов.
Измеряли оптическую плотность анализируемых растворов, модельных растворов и раствора сравнения по 3 раза с выниманием кюветы при длине волны 262 нм на спектрофотометре СФ-46 в кювете с толщиной слоя 10 мм. В качестве компенсационного раствора использовали 0,1 моль/л раствор кислоты хлористоводородной.
Результаты исследования и их обсуждение. Ранее [4] нами были предложены следующие критерии и процедура оценки приемлемости прецизионности УФ- спектрофотометрических методик количественного определения аналитов в биологических жидкостях, применяемых в судебно-токсикологическом анализе:
- применение нормализованных координат;
- подтверждение прецизионности методики проводят в двух направлениях - на модельных растворах (без матрицы) и на образцах матрицы;
- проверку прецизионности методики по модельным растворам проводят путем расчета их концентрации с использованием соответствующей линейной
model
зависимости; полученные значения X calc , %
используют для расчета
model
RR , % и
model
sa mp le ;
∆
model
sample предложены критерии
приемлемости в рамках двух подходов, основанных на: 1) предположении равенства неопределенности процедуры пробоподготовки и неопределенности количественного определения аналита в модельных ∆ model
sample ≤ 10,00%); 2) предположении
незначимости неопределенности количественного model sample ≤ 4,52%);
- оценку прецизионности методики на образцах матрицы проводят на двух уровнях - within-run и between-run - с использованием калибровочных и модельных образцов;
- определение within-run прецизионности проводят путем расчета концентрации калибровочных образцов для каждой последовательности по индивидуальным значениям оптической плотности с использованием линейной зависимости, полученной для данной последовательности; полученные значения Xcalc, % используют для расчета RR, % и Δsample, для оценки которой предложен следующий критерий приемлемости:
≤ max∆ ‚ = 0,707 · max ∆. = 14,14%;
sa mp le sa mp le As
• определение between-run прецизионности проводят в три этапа - рассчитывая разность между средними RR, % значениями
в разные дни (она не должна превышать 4,52%), а также рассчитывая концентрации модельных образцов и средние концентрации калибровочных образцов с использованием линейной зависимости, полученной по средним значениям параллельных последовательностей;
- исследования модельных образцов проводят для трех параллельных последовательностей/run, образцы биологической матрицы для которых получены из различных источников; для D = 25 - 125% каждая последовательность состоит из 3 модельных образцов (концентрации соответствуют точкам 25%, 50% и 100% в нормализованных координатах), для D = 25 - 150% и 25 - 175% - из 4 образцов (концентрации соответствуют точкам 25%, 50% и 150% или 175% соответственно в нормализованных координатах);
- полученные значения Xcalc, % для модельных и калибровочных образцов используют для расчета RR, %, intra
а также величин sample и Δsample, для оценки которых intra предложены критерии приемлемости ( sample ≤ 20,00% и Δsample ≤ 14,14%).
Для иллюстрации предложенных подходов к определению и оценке прецизионности использовали УФ- спектрофотометрическую методику количественного определения доксиламина в крови [3]; за 100% принимали летальную концентрацию доксиламина в крови [5] - 25 мг/л (что соответствует 36 мг/л доксиламина сукцината).
В табл. 1 приведены результаты измерений значений оптической плотности модельных растворов, рассчитанные значения концентраций модельных растворов и величины RR, % для различных диапазонов применения методики. Данные model таблицы 1 относительно величины sample , % свидетельствуют, что требования к сходимости результатов выдерживаются и для Подхода 1, и для Подхода 2.
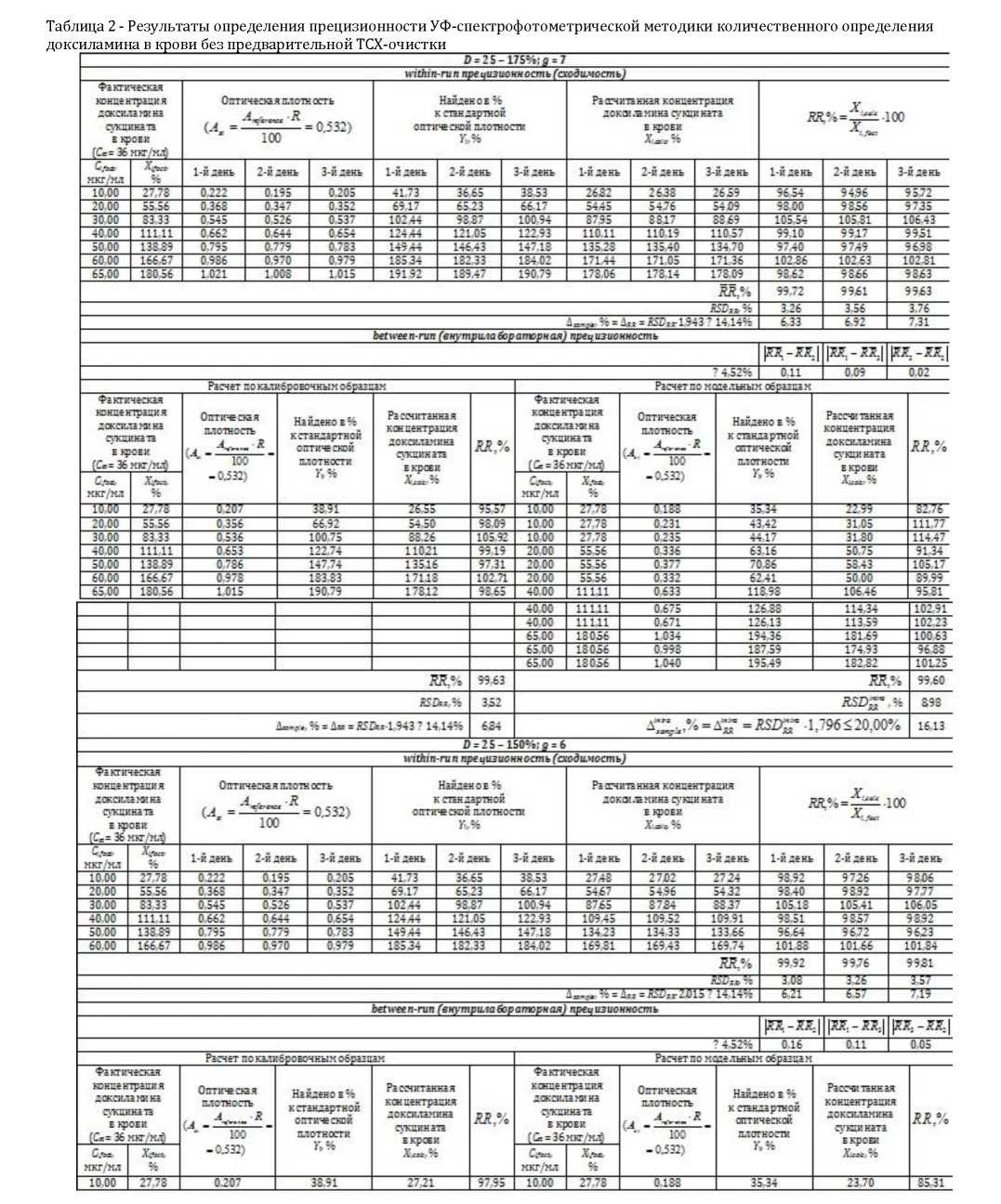
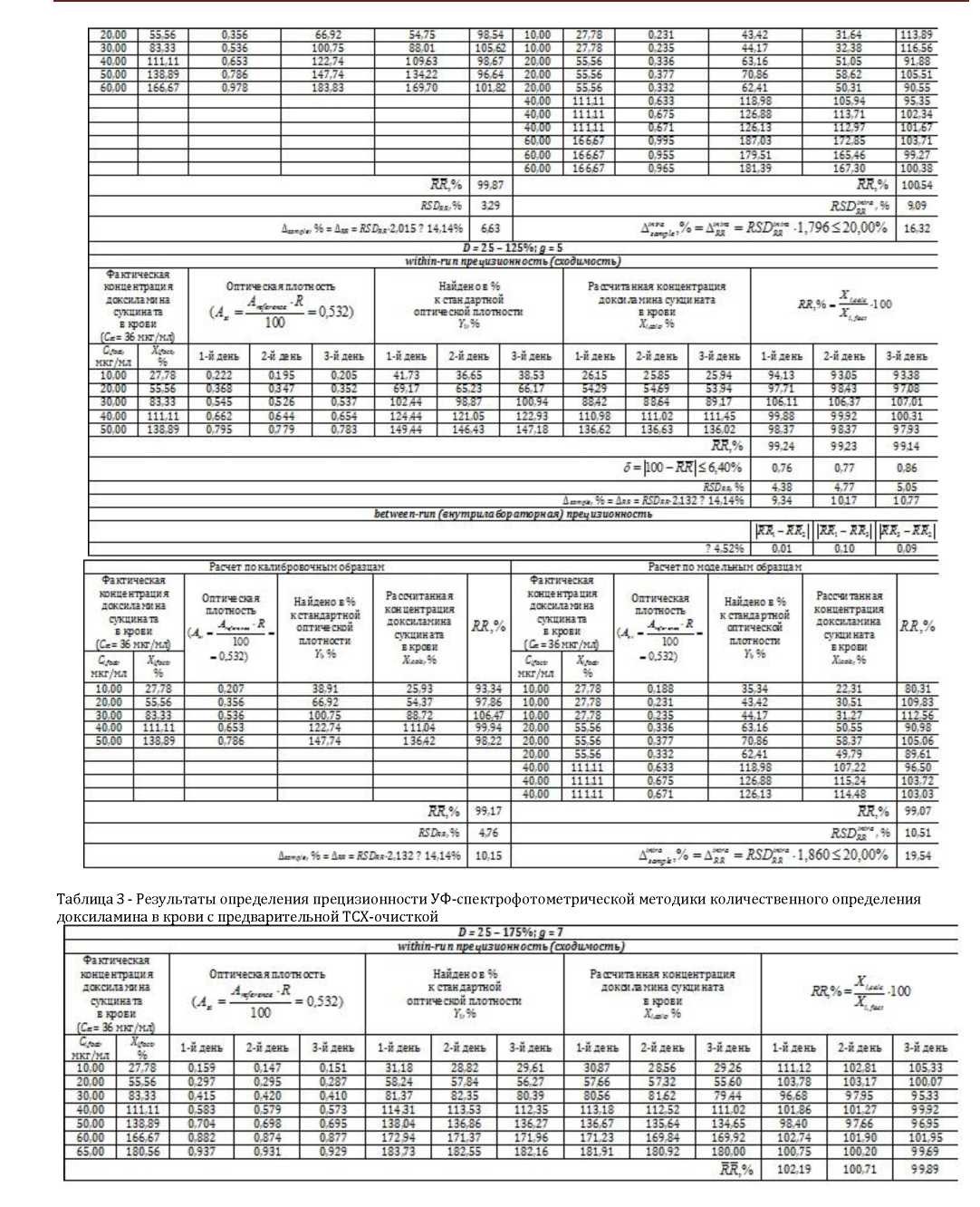
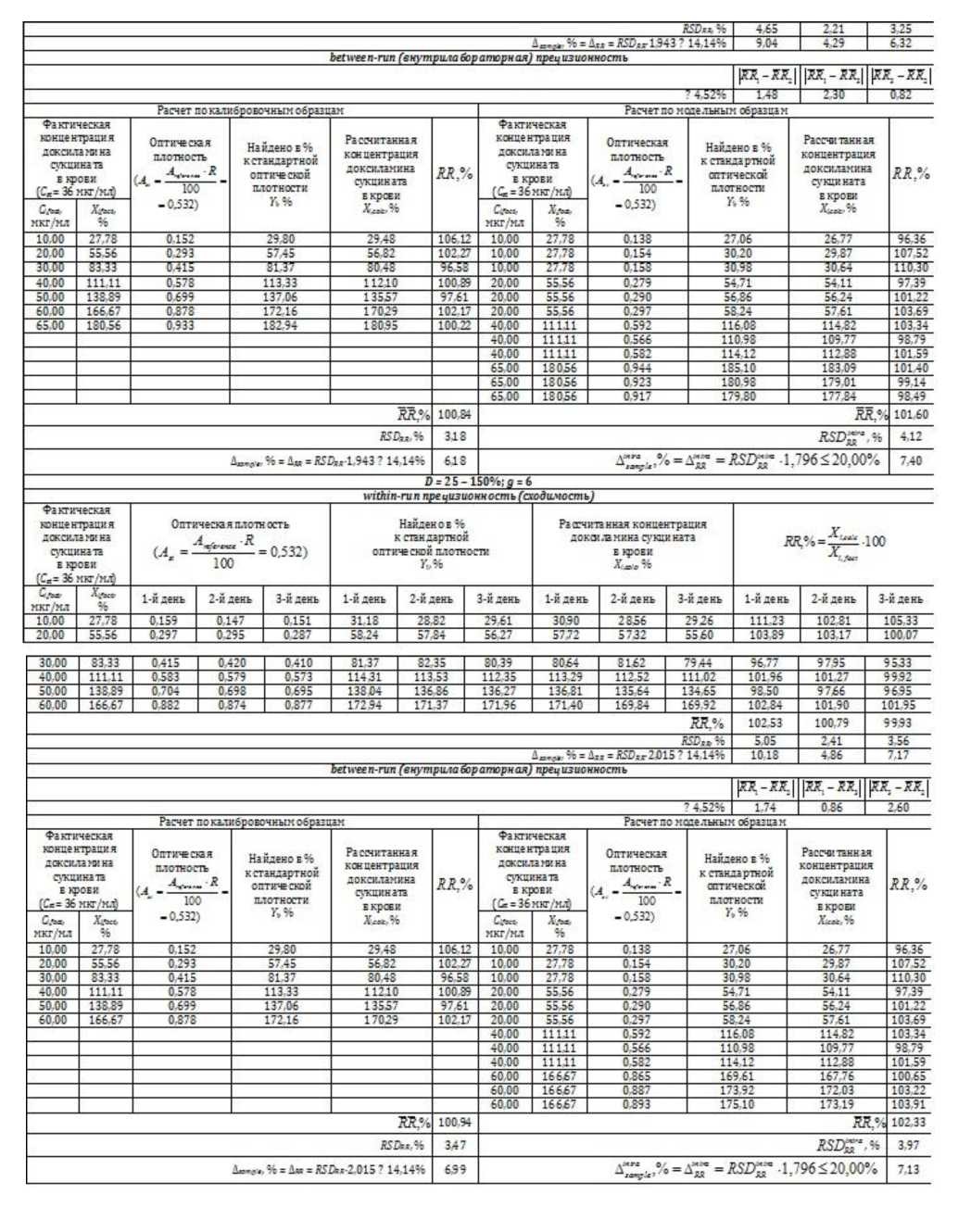
В таблице 2 - 3 приведены результаты измерений значений оптической плотности для калибровочных и модельных образцов, соответствующие им значения Xcalc, % и величины RR, % для различных диапазонов применения методики. Из приведенных в табл. 2 - 3 данных видно, что требования к сходимости и внутрилабораторной прецизионности выполняются для всех предложенных вариантов диапазонов применения методики и для обоих вариантов методики - с ТСХ-очисткой и без нее.
Для диапазона применения 25 - 125% в случае выполнения анализа без предварительной ТСХ-очистки величина intra sample достигает критических величин - 19,54%, поэтому для данного варианта методики лучше пользоваться более широким диапазоном применения.
Необходимо отметить, что в целом методики с выполнением предварительной ТСХ-очистки обладают лучшей внутрилабораторной прецизионностью и худшей сходимостью, чем методики без ТСХ-очистки, что объясняется введением дополнительных стадий пробоподготовки, естественным образом ухудшающих within-run прецизионность (оценивающую, в первую очередь, случайную ошибку, вносимую пробоподготовкой), но нивелирующих влияние смены источника происхождения матрицы на результаты анализа за счет более качественной очистки пробы (и тем самым улучшающих between-run прецизионность).
Выводы. Подходы к процедуре определения и оценке приемлемости прецизионности УФ-спектрофотометрических методик количественного определения аналитов в биологических жидкостях, применяемых в судебнотоксикологическом анализе, предложенные в нашей предыдущей работе [4], апробированы на примере УФ- спектрофотометрической методики количественного определения доксиламина в крови. Полученные результаты показали адекватность сформированных подходов.
Таблица 1 - Результаты определения прецизионности УФ-спектрофотометрической методики количественного определения доксиламина сукцината по модельным растворам
|
Фактическая концентрация доксиламина сукцината в модельном растворе (Ģ¡ţ = 28,8 мкг/мл) |
Оптическая плотность (.%- = 0,801) |
Найдено в % к стандартной оптической плотности model γi , % |
Рассчитанная концентрация доксиламина сукцината в модельном model растворе X i,calc , % |
model RR %〇 = i,calC ·1〇〇 γ- model X i , fa ct |
|
|
model Ci , fa ct , мкг/мл |
model X i , fa ct , % |
||||
|
D = 25 - 175% (g = 7) |
|||||
|
8,00 |
27,78 |
0,226 |
28,21 |
28,35 |
102,05 |
|
16,00 |
55,56 |
0,444 |
55,43 |
55,70 |
100,26 |
|
24,00 |
83,33 |
0,657 |
82,02 |
82,42 |
98,91 |
|
32,00 |
111,11 |
0,890 |
111,11 |
111,66 |
100,49 |
|
40,00 |
138,89 |
1,121 |
139,95 |
140,64 |
101,26 |
|
48,00 |
166,67 |
1,348 |
168,29 |
169,12 |
101,47 |
|
52,00 |
180,56 |
1,421 |
177,40 |
178,27 |
98,73 |
|
RR model % |
100,45 |
||||
|
RSDR°del,l⅛ |
1,27 |
||||
|
∖ ∖ -'% = x = RSDRRdel · 1,943 |
подход 1 |
≤ 10,00% |
2,47 |
||
|
подход 2 |
≤ 4,52% |
||||
|
D = 25 - 150% (g = 6) |
|||||
|
8,00 |
27,78 |
0,226 |
28,21 |
27,90 |
100,43 |
|
16,00 |
55,56 |
0,444 |
55,43 |
54,82 |
98,67 |
|
24,00 |
83,33 |
0,657 |
82,02 |
81,12 |
97,34 |
|
32,00 |
111,11 |
0,890 |
111,11 |
109,89 |
98,90 |
|
40,00 |
138,89 |
1,121 |
139,95 |
138,41 |
99,65 |
|
48,00 |
166,67 |
1,348 |
168,29 |
166,44 |
99,86 |
|
RR model % |
99,14 |
||||
|
RSDR°del,l⅛ |
1,09 |
||||
|
NmadR1le,% = XRRel = RSDRRdel · 2,015 |
подход 1 |
≤ 10,00% |
2,20 |
||
|
подход 2 |
≤ 4,52% |
||||
|
D = 25 - 125% (g = 5) |
|||||
|
8,00 |
27,78 |
0,226 |
28,21 |
28,07 |
101,04 |
|
16,00 |
55,56 |
0,444 |
55,43 |
55,15 |
99,27 |
|
24,00 |
83,33 |
0,657 |
82,02 |
81,61 |
97,94 |
|
32,00 |
111,11 |
0,890 |
111,11 |
110,56 |
99,50 |
|
40,00 |
138,89 |
1,121 |
139,95 |
139,25 |
100,26 |
|
RR model % |
99,60 |
||||
|
RSDR°dd,,⅛ |
1,16 |
||||
|
ă model q/ tmodel τ)C∣τ∖model ɔ 1 ɔ-ɔ ∖ample,% = RSDrr · 2,132 |
подход 1 |
≤ 10,00% |
2,47 |
||
|
подход 2 |
≤ 4,52% |
||||
СПИСОК ЛИТЕРАТУРЫ
- Клименко, Л. Ю. Анализ подходов к определению специфичности / селективности при проведении валидации аналитических методик в судебно-токсикологическом анализе. // Укр. мед. альм. – 2013. – Т. 16. - №1. – С. 47 – 49.
- Клименко, Л. Ю. Подходы к определению специфичности / селективности при валидации УФ-спектрофотометрических методик количественного определения в судебно-токсикологическом анализе / Л. Ю. Клименко, Г. П. Петюнин, Т. А. Костина // Фармация Казахстана. – 2013. – №8. – С. 53 – 56.
- Л. Ю. Клименко, С. Н. Трут, Г. П. Петюнин, И. М. Иванчук Модификация и валидация УФ-спектрофотометрической методики количественного определения доксиламина в крови: специфичность // Укр. журн. клін. та лаборатор. медицини. – 2013. – Т. 8. - №4. – С. 191 – 199.
- Klimenko, L. Yu. Approaches to determination of precision for UV-spectrophotometric methods of quantitative determination in forensic and toxicological analysis // Фармация Казахстана. – 2014. – №3. – С. 43 – 50.
- A. C. Moffat, M. D. Osselton, B. Widdop Clarke's analysis of drugs and poisons in pharmaceuticals, body fluids and postmortem material. – London: Pharmaceutical Press, 2011. – 2609 p.
- L. Yu. Klimenko, S. M. Trut, G. P. Petyunin, I. M. Ivanchuk Validation of UV-spectrophotometric methods of quantitative determination in forensic and toxicological analysis: recovery // Фармация Казахстана. – 2013. – №12. – С. 42 – 48.
- Klimenko, L. Yu. Development of approaches to validation of UV-spectrophotometric methods of quantitative determination in forensic and toxicological analysis: linearity and range // Фармацевтичний часопис. – 2014. – №1 (30). – С. 41 – 48.