Дефекты гена KCJN11, кодирующего Kir6.2 субъединицу калиевого канала, является наиболее частой причиной диабета, развивающегося до 6-месячного возраста. Гетерозиготная миссенс мутация в гене KCNJ11 вызывает нарушение биосинтеза инсулина и манифестацию СД в течение первых недель жизни ребенка. Впервые в Казахстане проведено молекулярно-генетическое исследование и описание 2 случаев неонатального СД. Показана роль молекулярно-генетического анализа дефектов бета-клеточной функции в дифференциальной диагностике сахарного диабета у детей первого года жизни.
Сахарный диабет (СД) у детей первого года жизни представлен гетерогенной группой заболеваний, включающей как СД 1 типа в результате аутоиммунного поражения поджелудочной железы, так и ряд моногенных форм, ассоциированных с мутациями в генах, обеспечивающих нормальное развитие и функцию панкреатических бета-клеток. К моногенным заболеваниям относят болезни, вызванные мутацией единственного гена. Согласно этиологической классификации СД (ВОЗ, 1999) моногенные формы СД относят к группе специфических типов диабета, связанных с генетическими дефектами β-клеточной функции и включают диабет типа MODY и неонатальный СД (НСД) [1,2].
Для неонатального СД характерно изолированное нарушение глюкозостимулироованной секреции инсулина (мутации в генах KCNJ11, ABCC8, GCK, INS) [3-5]. В ряде случаев СД, дебютировавший на первом году жизни ребенка, является частью таких синдромов как синдром Дауна, Клайнфельтера, Тернера, Вольфрама, Лоуренса–Муна–Барде–Бидля, Прадера–Вилли [1].
Выделяют две формы заболевания – транзиторный неонатальный диабет (ТНСД) и перманентный неонатальный СД (ПНСД). При дифференциальном диагнозе транзиторного и перманентного диабета вследствие нарушения созревания фетальной возникают большие трудности. В обоих случаях поджелудочной железы (таблица 1) [3-5].
имеет место недостаточность секреции инсулина
Таблица 1 - Формы и частота мутаций генов неонатального СД
|
Транзиторный НСД |
Перманентный НСД |
Синдромы и панкреатическая аплазия |
|
6qZAC (71%) |
INS (16%) |
FOXP3, HNF-1β, IPF1 |
|
Kir6.2 (11%) |
Kir6.2 (29%) |
|
|
SUR1 (11%) |
SUR1 (12%) |
|
|
GCK (3%) |
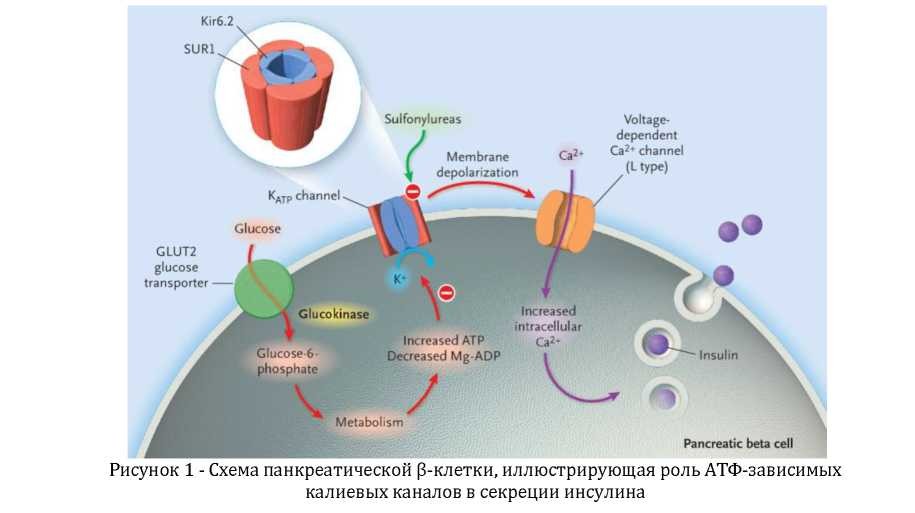
Для понимания патогенеза неонатального СД необходимо знать физиологию бета-клетки. Калиевые каналы ß-клеток глубоко вовлечены в регулирование секреции инсулина (рис. 1). Калиевые каналы – это октомерный комплекс, состоящий из 4 внутренних ректификационных калиевых каналов (Kir6.2) и четырех субъединиц рецепторов к сульфонилмочевине (SUR1). Kir6. 2 связываясь с ATΦ, способствует закрытию калиевого канала, а магниевые нуклеотиды связываются с SUR1, вызывая активацию канала. После пищевой нагрузки наблюдается повышение внутриклеточного содержания глюкозы, сопровождающееся образованием АTФ и закрытием калиевых каналов. Данный процесс приводит к деполяризации мембраны клетки и выходу накопившегося инсулина. Голодание же снижает внутриклеточное соотношение ATФ/AДФ, что приводит к открытию калиевых каналов и торможению секреции инсулина (рисунок 1).
Ген KCJN11 кодирует Kir6.2 субъединицу, а ген ABCC8 – субъединицу SUR1. И Kir6.2, и SUR1 жизненно необходимы для правильного регулирования секреции инсулина. Активирующая мутация в KCNJ11, вызывая закрытие АТФ-зависимых калиевых каналов, ведет к развитию неонатального СД. Патогенез заболевания связан с уменьшением выхода калия из клетки и гиперполяризацией мембраны β- клетки, сопровождающихся сокращением секреции инсулина. У детей с данной мутацией обычно развивается диабет в возрасте до 6 месяцев, сопровождающийся кетоацидозом и отсутствием C- пептида. Приблизительно 25% детей с транзиторным неонатальным диабетом также имеют активирующие мутации в KCNJ11, и иногда диабет у таких детей возникает позже 6-месячного возраста без наличия неонатального диабета в анамнезе. Таким образом, гетерозиготная активирующая мутация в KCJN11 гене, кодирующая Kir6.2 субъединицу калиевого канала, является наиболее частой причиной как транзиторного, так и перманентного неонатального диабета, развивающегося до 6-месячного возраста [6,7].
У детей с активирующей мутацией в KCNJ11 наблюдается хорошая ответная секреция инсулина на препараты сульфонилмочевины, которые связываются с SUR1 на АТФ-зависимых калиевых каналах и закрывают их через ATФ-независимый путь. Имеются сообщения, что до 90% детей с НСД, вызванным мутацией в KCNJ11, могут быть успешно переведены с инсулина на сульфонилмочевинные препараты с улучшением гликемического контроля независимо от длительности СД [5,6]. Поэтому у всех детей с манифестацией диабета до 6-месячного возраста необходимо проводить генетическое тестирование. Дополнительным поводом для проведения исследования мутации KCNJ11 является семейный анамнез со случаями диабета, возникшего
в раннем возрасте, хотя некоторые дети имеют мутации de novo. Как только ребенок будет стабилизирован на инсулинотерапии, а 1<¡г6.2-диабет установлен, может быть предпринята попытка перевода на таблетированные препараты из группы сульфонилмочевины.
Цель: Молекулярно-генетическая диагностика моногенных форм СД в Казахстане до настоящего времени не проводилась. Нами впервые проведено подобное исследование у детей с манифестацией СД на первом году жизни.
Материалы и методы
В исследование включены 4 пациента с манифестацией СД на первом году жизни, а также проведен забор крови и последующий молекулярногенетический анализ у обоих их родителей. У двоих детей дебют СД отмечен в возрасте 1 мес, у одного – в 6 мес, у последнего – в возрасте 10 мес. Средний возраст пациентов на момент проведения молекулярно-генетического анализа составил 1,3 года (0,5-2,5 года). Среди обследованных детей было 3 девочки и 1 мальчик.
Молекулярно-генетические исследования.
Проводился забор цельной крови в пробирки с ЭДТА и последующее выделение ДНК из периферических лейкоцитов с использованием стандартных методов. С помощью ПЦР амплифицировали фрагмент геномной ДНК, охватывающие кодирующую последовательность генов KCNJ11, INS, ABCC8. Молекулярно-генетическое исследование данных генов осуществлено в генетической лаборатории Университета Экзетера (Великобритания).
Результаты исследования
У 2 из 4 детей с манифестацией СД в возрасте 4 нед были обнаружены гетерозиготные миссенс мутации в гене KCNJ11, экзон 1 – c.685G>A в гене KCNJ11, экзон 1, с.602>Æ В одном случае мутация в гене KCNJ11 возникла de novo, тогда как в другом случае заболевание имело семейный характер (мать в возрасте 25 лет и дочь с мутацией в гене KCNJ11, экзон 1 – c.685G>A).
У остальных двух детей с манифестацией СД в возрасте 6 и 10 мес дефекты в генах KCNJ11, INS, ABCC8 обнаружены не были. Кроме того у этих пациентов определялись высокие титры антител к островковым клетка поджелудочной железы.
До генетической верификации у одного ребенка с семейным характером СД был диагностирован транзиторный неонатальный СД. Инсулинотерапия у ребенка была полностью отменена. В данном случае молекулярно-генетическое исследование выявило СД у матери, у которой заболевание протекало бессимптомно. Матери ребенка была назначена терапия препаратом сульфонилмочевины гликлазид в дозе 60 мг.
Проведение молекулярно-генетического анализа и выявление мутации в гене KCNJ11 позволило другому ребенку с перманентным неонатальным СД отменить интенсифицированную инсулинотерапию и назначить таблетированный препарат сульфонилмочевины глибенкламид в дозе 7,5 мг/день.
В остальных двух случаях у детей с манифестацией СД в возрасте 6 и 10 мес, у которых молекулярногенетический анализ не выявил каких-либо мутаций в генах KCNJ11, INS, ABCC8, диагностирован СД 1 типа и продолжена интенсифицированная инсулинотерапия.
Ниже приводим описание собственного наблюдения детей с неонатальным диабетом, ассоциированным с гетерозиготными миссенс мутациями в гене KCNJ11 . Ребенок К.А., 2 лет, находится под наблюдением в РДКБ «Аксай». В возрасте 1 мес поступила в инфекционную больницу с жалобами на повышение температуры тела до 38 градусов, жажду, полидипсию, полиурию, потерю в весе, сухость кожных покровов, слабость. При поступлении выявлена гипергликемия 30 ммоль/л, кетонурия. Был диагностирован сахарный диабет 1 типа, проведена внутривенная регидратационная терапия и инсулинотерапия. Через 5 дней девочка была переведена в ДГКБ №2, где при поступлении отмечалась заторможенность, сонливость ребенка. При обследовании выявлена гипогликемия 2,5 ммоль/л, в связи с чем была проведена внутривенная инфузия 40% глюкозы. Далее ребенку проводилось подкожное введение актрапида по 1 ед 5 раз перед каждым кормлением грудным молоком. В дальнейшем ребенок был переведен на режим многократных инъекций: 2 ед лантуса перед сном и новорапид по 1 ед перед приемом пищи.
При обследовании матери, которой исполнилось 23 года, была выявлена гипергликемия 15 ммоль/л. У нее был диагностирован сахарный диабет 2 типа и назначена сахароснижающая терапия метформином 850 мг и 30 мг гликлазида (препарат сульфонилмочевины) утром. Однако у матери не отмечался избыточный вес и отсутствовали симптомы инсулинорезистентности (acanthosis nigricans, атерогенная дислипидемия).
В возрасте 2 мес у ребенка отмечалась склонность к гипогликемии, которая была расценена как «медовый месяц», в связи с чем дозы инсулина были снижены.
В возрасте 1,5 лет ребенок был госпитализирован в РДКБ «Аксай». Рост составил 80 см, вес - 11 кг. Перед поступлением в стационар получала инсулинотерапию: 1 ед протафана перед сном и 0,2 ед новорапида перед 3 основными приемами пищи. Обращало внимание, что при обследовании отсутствовали симптомы дефицита инсулина: кожа и слизистые были хорошо увлажнены, не было дефицита массы тела, полиурии и полидипсии. Гликемический профиль от 08.05.12 08.00-
5,9ммоль/л, 11.00-6,9ммоль/л, 13.00- 5,2ммоль/л, 15.00- 6,6ммоль/л, 18.00- 5,2ммоль/л, 20.00- 6,6 ммоль/л, 22.00- 5,3 ммоль/л, 03.00-4,7 ммоль/л. Уровень гликированного гемоглобина (НВА1с) был низким 3, 75% (норма лаборатории 4,2-5,7%).
Учитывая вышесказанное, диагноз сахарного диабета 1 типа был отвергнут на основании возраста манифестации диабета (1 мес), низкой потребности в инсулине (менее 1 ед в сутки), низкого уровня гликированного гемоглобина (НвА1с 3,75%), свидетельствующего о скрытых гипогликемиях, низкой массы при рождении (2800 гр, несмотря на то, что у матери был не диагностированный бессимптомный сахарный диабет). В связи с этим выставлен диагноз транзиторного неонатального диабета и ребенку была отменена инсулинотерапия. Кроме того, учитывая семейный характер заболевания, молодой возраст (23 года), отсутствие признаков инсулинорезистентности, у матери ребенка отвергнут диагноз сахарного диабета 2 типа и заподозрен моногенный СД диабет. Матери был отменен метформин, лечение продолжено гликлазидом в дозе 60 мг, была достигнута нормогликемия. В настоящее время ребенок находится под нашим наблюдением, сахароснижающую терапию не получает, гликемия в норме.
У данного ребенка в возрасте 2,5 лет и обоих родителей проведено молекулярно-генетическое исследование генов KCN111, INS и ABCC8. У ребенка и ее матери выявлена одинаковая гетерозиготная миссенс мутация в гене KCN111, экзон 1 – c.685G>A, подтверждающая диагноз транзиторного неонатального СД. У матери СД протекал бессимптомно, был выявлен при манифестации СД у ее ребенка, подтвержден семейный характер диабета при молекулярно-генетическом анализе.
Приводим наше собственное наблюдение за другим ребенком с перманентным неонатальным СД.
Ребенок С.А., 2 мес, госпитализирован в возрасте 1 мес в реанимационное отделение по месту жительства с высокой температурой, жаждой, полиурией, полидипсией, потерей веса. При обследовании выявлена гипергликемия 32 ммоль/л, кетонурии не отмечалось. Учитывая возраст ребенка (1 мес), отсутствие кетоацидоза был диагностирован неонатальный сахарный диабет. Проводилась регидратация, инсулинотерапия лантусом 2 ед/сутки и 0,1 ед хумалога перед кормлением грудью. На фоне инсулинотерапии у ребенка отмечалась сонливость, слабость, ребенок плохо сосал грудь (отмечалась гипогликемия 2,7 ммоль/л).
Для дальнейшего лечения пациент был направлен в ДГКБ№2, где впервые в Казахстане ребенок был переведен с инсулинотерапии на лечение манинилом (глибенкламид, таблетированный препарат сульфонилмочевины) в дозе 0,6 мг/кг/сутки перед едой. На данной терапии была достигнута нормогликемия.
Ребенку в возрасте 1 года также проведено молекулярно-генетическое исследование генов KCN111, INS and ABCC8. Выявлена гетерозиготная миссенс мутация в гене KCN111, экзон 1, с.602>Æ У родителей при обследовании никаких мутаций не обнаружено, что свидетельствует о наличии у ребенка мутации de novo. Выявление мутации в Kir6.2 субъединице подтвердило обоснованность назначения терапии препаратом сульфонилмочевины (глибенкламид) и отмену инсулинотерапии у данного ребенка.
Наш личный опыт подтверждает, что гетерозиготная активирующая мутация в KC1N11 гене, кодирующая Kir6.2 субъединицу калиевого канала, является наиболее частой причиной диабета, развивающегося до 6-месячного возраста.
Кроме этих двух детей, у которых неонатальный СД диагностирован в возрасте 1 месяцев, нами были обследованы дети, у которых СД манифестировал в возрасте 6 и 10 месяц. Молекулярно-генетическое исследование не выявило у этих детей каких-либо мутаций а генах 6qZAC, КСÑĴ11, ABCC8, ответственных за неонатальный СД. Таким образом, отсутствие мутаций в данных генах свидетельствует в пользу наличия у этих детей аутоиммунного СД 1 типа. При дальнейшем обследовании у детей выявлены антитела к островковым клеткам поджелудочной железы.
Таким образом, диагностика моногенных форм сахарного диабета в Казахстане до сегодняшнего дня не проводилась. Нами впервые диагностированы случаи неонатального СД на основании молекулярногенетического анализа дефектов бета-клеточной функции у детей первого года жизни, позволившего провести дифференциальную диагностику СД у детей с манифестацией заболевания на первом году жизни. Молекулярно-генетический анализ выявил вероятность неонатального СД в случае манифестации СД в возрасте до 6 мес. При манифестации СД в более поздние сроки (после 6 мес жизни) вероятнее аутоиммунный характер СД 1 типа. Выводы:
Диагностика моногенных форм диабета имеет огромное научное значение, поскольку они являются природной моделью, позволяющей изучить физиологию бета-клеточной функции, основные механизмы развития нарушений углеводного обмена. Молекулярно-генетическое исследование детей первого года жизни позволяет провести дифференциальную диагностику неонатального СД и СД 1 типа.
Выявление гетерозиготной активирующей мутации в KC1N11 гене позволяет в случае транзиторного неонатального СД отменить инсулинотерапию, а при перманентной форме – перевести ребенка на лечение таблетированным препаратом сульфонилмочевины, отменив множественные инъекции инсулина.
СПИСОК ЛИТЕРАТУРЫ
- Дедов И.И., Кураева Т.Л., Петеркова В.А. Сахарный диабет у детей и подростков. – М.: 2007. - С.135-156.
- American Diabetes Association: Diagnosis and Classification of Diabetes (Position Statement) //Diabetes Care. – 2014. –
- 33 (Suppl.1). - Р. 62-69.
- Barret T.G.//Differential diagnosis of type 1 diabetes: which genetic syndromes need to be considered // Pediatric Diabetes. - 2007. - № 8. - P. 15-23.
- Fajans S.S., Bell G.I. Phenotypic heterogeneity between different mutations of MODY subtypes and within MODY pedigrees // Diabetologia. – 2006. – №49. – Р.1106–1108.
- Ellard S., BellanM-Chantelot C., Hattersley A.T. European Molecular Genetics Quality Network (EMQN) MODY group. Best practice guidelines for the molecular genetic diagnosis of maturity-onset diabetes of the young // Diabetologia. – 2008. – №51. – Р.546–553
- Anna L. Gloyn, D.Phil., Ewan R. Pearson M.R. et al. Activating Mutations in the Gene Encoding the ATP-Sensitive Potassium-Channel Subunit Kir6.2 and Permanent Neonatal Diabetes // NE1M. – 2004. – №18. - P. 1828-1849.
- Oscar Rubio-Cabezasa, Sarah E.Flanagana, Annet Damhuisd, Andrew T. Hattersleya. K+ATP channel mutations in infants with permanent diabetes diagnosed after 6 months of life // Pediatric Diabetes. – 2011. - Р.1399-5448.