Type 2 diabetes mellitus is a complex metabolic disorder characterized by a relative deficiency of insulin in the presence of hepatic, adipose tissue, and skeletal muscle insulin resistance. The pathological process underlying the ß-cell dysfunction occurs already prior to the disease onset. While at the initial stage, ß-cell mass and insulin secretory function are sufficiently well maintained in the majority of individuals with type 2 diabetes, the later stages are characterized by aggravating insulin deficiency. The clinical course of the disease requires escalating therapy with oral drugs over time and eventually consistent application of insulin at the late stage for control of glycemia. Oral therapies are quite effective in improving the short-term insulin secretory capacity, but are incapable of preventing the inexorable decline in ß-cell function during diabetes progression. On the other hand, long-term use of antidiabetic agents is not without various side effects. Since a series of clinical trials have recently shown that implementation of short-term intensive insulin therapy in individuals with newly diagnosed type 2 diabetes can drastically improve and preserve ß-cell function and induce glycemic remission, this treatment strategy has gained considerable interest. However, whether early intensive treatment with insulin can really provide longer-term protection of the pancreatic ß-cells and may be preferable to other therapy modalities is a question that is not yet clearly established and requires appropriate clinical studies.
Diabetes mellitus is a complex metabolic disorder that is characterized by absolute (type 1 diabetes) or relative (type 2 diabetes) deficiency of insulin. Autoimmune-mediated processes trigger dysfunction and early destruction of pancreatic ß-cells in type 1 diabetes, whereas gradual reduction in ß-cell mass, defective insulin secretion and sensitivity are the main factors causing initiation and progression of type 2 diabetes. An early indication of the failing ß-cell is the progressive deterioration of glucoregulation with excessive glucose excursion after carbohydrate ingestion and the sequential occurrence of sustained chronic hyperglycemia at postprandial times and during fasting periods. By the time the disease is clinically diagnosed, ß-cell mass and ß-cell function have declined by 25–60 % [1]. Early functional alterations of ß-cells in individuals with type 2 diabetes characteristically include reduced or absent first-phase insulin/C-peptide response to glucose and blunted or delayed insulin/C-peptide release during a mixed-meal test. Chronic sustained hyperglycemia has been shown to exert deleterious effects on the ß-cells via several pathological pathways, among which apoptosis induced by glucotoxicity is the most harmful lesion. ß-Cell function, i.e. glycemic control, declines more rapidly in poorly controlled than in well-controlled diabetes, as has been documented in the Diabetes Control and Complication Trial (DCCT/EDIC) [2] and the United Kingdom Prospective Diabetes Study (UKPDS) [3]. On the other hand, these trials demonstrated that intensive therapy creates a metabolic memory that slows down the development of diabetic complications. The question arises whether strict glycemic control early in the history of dysglycemia is able to normalize or at least preserve the residual ß-cell function over longer times. A wealth of data from numerous experimental and clinical studies has suggested that reducing any hyperglycemic stress, either induced by sustained chronic hyperglycemia or excessive glucose variability, leads to amelioration of the ß-cell function [4]. These data provide hope that the factors mediating ß-cell preservation can eventually be identified.
ß-Cell failure in type 2 diabetes
The functional and structural alterations of ß-cells in type 2 diabetes are progressive in nature. Mild postprandial hyperglycemia is the earliest metabolic defect observed in individuals with impaired glucose tolerance. It is of note that, at this stage, the decline in ß-cell function is associated with little increase in peripheral insulin resistance, as reported by Kahn [5] and Gastadelli et al. [6]. As disturbance of glucose home ostasis progresses, postprandial glucose excursions become excessive and prolonged, followed by hypersecretion of insulin to compensate for peripheral insulin resistance. These prandial abnormalities in glucose regulation indicate a deficit in first-phase insulin secretion, which gives rise to insufficient suppression of hepatic glucose production. The resulting hyperglycemia launches a pathological process in which both ß-cell mass and function are greatly lost and insulin substitution becomes unavoidable for patients´ survival. Interestingly, the rapidity of the functional decline depends on a variety of environmental as well as genetic factors. A summary of the potentially most important factors to be involved in ß-cell dysfunction is shown in Figure 1.

Figure 1. Synopsis of potentially negative factors contributing to ß-cell dysfunction in type 2 diabetes
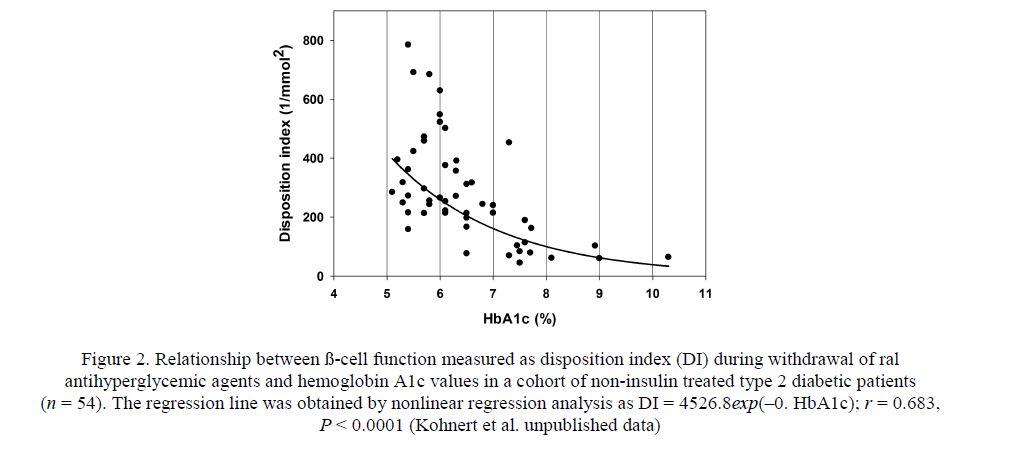
Figure 2. Relationship between ß-cell function measured as disposition index (DI) during withdrawal of ral antihyperglycemic agents and hemoglobin A1c values in a cohort of non-insulin treated type 2 diabetic patients (n = 54). The regression line was obtained by nonlinear regression analysis as DI = 4526.8exp(–0. HbA1c); r = 0.683, P < 0.0001 (Kohnert et al. unpublished data)
According to current knowledge, there is no doubt that hyperglycemia and excessive fluctuation in glucose levels are main contributors to ß-cell failure [7]. Figure 2 shows that ß-cell function (Disposition index) declines in an exponential manner with worsening long-term glucose control (HbA1c), and patients with HbA1c levels in the range of 5.0–6.5 % maintain better preserved ß-cell function than those above this range, as indicated by higher disposition index values. It should be noted that the disposition index is the most comprehensive measure to express ß-cell function, because it takes whole-body insulin resistance into account.
The pathway from sustained chronic hyperglycemia, increased glycemic variability, and elevated nonesterified fatty acids to ß-cell damage includes glucolipotoxicity, generation of oxidative stress and nitrosative stress through excessive production of reactive oxygen (ROS) and reactive nitrogen species (RNS), respectively [8]. The unbalanced formation of ROS and RNS species promotes lipid peroxidation, protein oxidation, mitochondrial and genomic DNA damage. Furthermore, the interference with signal transduction pathways can lead to ß-cell damage by a various mechanisms [9]. In contrast to animal experiments, evidence for the clinical importance of these processes in humans is so far lacking. Nevertheless, type 2 diabetic islets were shown to contain significantly higher concentrations of stress markers than pancreatic islets obtained from non-diabetic donors, suggesting a causal link between increased oxidative stress and decreased glucose-stimulated insulin secretion. The very low levels of intracellular antioxydant enzymes, such as catalase, glutathione peroxidase, and superoxide dismutase [10] may explain the high vulnerability against ROS and RNS and subsequent changes resulting in a variety of cellular dysfunctions and finally ß-cell apoptosis. Butler et al. [11] ascribed the mechanism responsible for reduced ß-cell volume to a 3to 10-fold increase in the rate of apoptosis, they observed in obese patients with type 2 diabetes as compared to lean nondiabetic subjects.
Genome-wide association studies identified several risk loci for type 2 diabetes [12], and it has been shown that some genetic risk variants (i.e., TCF7L2, KCNJ11, CDKAL1) act through perturbation of glucose-stimulated insulin secretion. CDKAL1, for example, is strongly expressed in the endoplasmic reticulum and Golgi apparatus in the ß-cell and may affect insulin secretion by causing stress and mitochondrial disruption due to misfolding or defective processing of proteins [13]. BMI-associated variants such as FTO were found to be implicated in regulation of lipid levels [14] and can thus modulate insulin resistance.
Insulin resistance is already established in individuals who are prone to develop type 2 diabetes but still having normal glucose tolerance. High fat diet and its metabolic consequences of increased body mass index are the critical factors in the development of insulin resistance. In order to compensate for the elevated insensitivity of skeletal muscle and liver, ß-cells are forced to hypersecrete insulin owing to chronic sustained hyperglycemia. This vicious cycle creates endoplasmic reticulum stress, probably triggering an apoptotic signal with subsequent destruction of ß-cells. It should be noted; however, that insulin resistance can also exist without ß-cell dysfunction.
Of the several mechanisms, which have been proposed to induce ß-cell failure in type 2 diabetes, various components of an inflammatory process are likely to be involved [15, 16]. The observation of amyloid deposits and fibrosis in pancreas section from patients with type 2 diabetes [17] is a strong indication for the occurrence of inflammatory processes in islets. Furthermore, it could be shown by Böni-Schnetzler et al. [18] that hyperglycemia increases interleukin-1ß production at the protein level, a factor that contributes to glucotoxicity. In the presence of elevated glucose concentrations, lipids have shown to exert deleterious effects on ß-cells, and cytokines (i.e., TNFα, interleukin-6, and leptin) secreted by fat cells may act directly or via activation of the innate immune system [19].
ß-Cell dysfunction can already originate in utero. Even though there are conflicting reports in the literature, several studies in small-for-gestational-age neonates demonstrated defects in glucose homeostasis [20]; and a study conducted by Nicolini et al. [21] in intrauterine growth restricted fetuses at 26–33 weeks of gestation found a complete absence of the first-phase insulin secretion, whereas Wang et al. [22] reported higher plasma insulin concentrations in small-forestational-age infants at 72 hours post partal. Indeed, the prevalence of type 2 diabetes is higher in individuals who had been exposed to intrauterine growth restriction during fetal development. Although the underlying mechanism for disturbances in glucose homeostasis occurring in small-for-gestational-age infants postnatal or later in life are not well understood, fetal under nutrition associated with placental insufficiency appears to be the primary cause.
Antidiabetic therapy and reversibility of ß-cell dysfunction
Although it has been shown that reversal of ß-cell failure and insulin resistance can be achieved without any antidiabetic medication, merely by dietary energy restriction [23], this treatment strategy requires longterm adherence and does not work in most individuals with type 2 diabetes. Thus pharmacologic intervention is inevitable to control hyperglycemia. Despite growing numbers of antidiabetic agents, the ideal drug that normalizes levels of glycemia throughout day and night is not yet available. In type 2 diabetes, a stepwise approach is routinely used to manage glycemic control. However, this approach has been questioned as it does not ensure consistently good glycemic control in the majority of patients. The «A Diabetes Outcome Progression Trial» (ADOPT) demonstrated, for example, that regardless of the oral drug initially used, monotherapy failed to a remarkable extent. The incidence of secondary therapy failure at five years was 34 %, 21 % and 15 % for glyburide, metformin, and rosiglitazone, respectively [24]. Two main conclusions can be derived from these outcomes: (1) the therapeutic approach did not address the pathological mechanisms, i.e., progressive declining ß-cell function and (2) monotherapy with oral drugs will surely fail at some time during disease advancement.
Glucagon-like peptide 1 (GLP-1) receptor agonists have shown promising positive effects on ß-cell function and even on ß-cell mass [25]. In a randomized clinical trial, Bunck et al. [26] investigated metabolic effects of exenatide in metformin-treated type 2 diabetic patients. Although these authors observed improvement of ß-cell function and glycemic control during one year of treatment, this effect was not sustained; ß-cell function and glycemia returned to pretreatment levels at 4 weeks after drug discontinuation. Moreover, in a recent, randomized controlled trial, Gudipaty and coauthors [27] evaluated the ß-cell secretory capacity of exenatide with glimepiride as a comparator early in the course of type 2 diabetes and found that the acute insulin response to arginine remained unchanged after 6 months of treatment with exenatide, whereas the sulfonylurea increased the ß-cell secretory capacity.
Analyzing the burden of treatment failure in type 2 diabetes, Brown et al. [28] came to the conclusion that treatment needs to be changed earlier and less likely to fail. As a consequence, antidiabetic therapy both capable of correcting the pathogenetic ß-cell abnormalities, as outlined by Del Prato and colleagues [29], and timely provided may ensure glycemic stability. The proposal by DeFronzo et al. [30] to initiate triple therapy with antidiabetic agents at the earliest stage of the disease is further reaching and in line with the multifactorial nature of type 2 diabetes, as outlined above in Fig 1. A strategy capturing several of the indicated factors at once, e.g., insulin resistance, inflammation, glucotoxicity, and lipotoxicity might be more efficacious than customary stepwise approaches to achieve long-term glycemic control. However, justification for implementation of such an early combination treatment requires large long-term clinical trials.
Initiation of early insulin therapy―potential benefits and negative consequences
The beneficial effect of insulin administration shortly after onset of diabetes has been demonstrated in experimental as well as clinical studies. For example, in pre-hyperglycemic animals and models for type 2 diabetes, insulin administration to normalize glycemia produced improvement of ß-cell secretory function and islet insulin content [31, 32]. In 1996, Kobayashi and colleagues [33] already reported that small doses of subcutaneous insulin prevented progressive ß-cell failure in patients with Latent Autoimmune Diabetes of Adults (LADA) over a follow-up of 30 months. Actually, insulin has been shown to be capable of preventing ß-cell defects [34] by reduction of glucolipotoxicity [35, 36] and inhibition of oxidative stress, as recently shown by Monnier et al. [37].
In the past ten years, several studies have shown that administration of insulin either shortly after onset or early in the course of the disease can improve both ß-cell secretory function and insulin resistance [38– 44]. Table summarizes trials that were published from 2003 to 2012 and in which ß-cell function has been assessed, using established methods. It is interesting that in some clinical trials in which initial treatments with insulin and oral hypoglycemic agents were compared head-to-head, both treatment modalities achieved comparable glycemic control. However, the drug-free remission with insulin exceeded by far that attained with oral drugs, for example, 62.5 vs. 0.5 %, at 12 months in the study performed by Chandra et al. [45], and similar studies also showed that ß-cell function parameters proved to be better preserved in the insulin than in the oral drug-treated groups [46, 47]. It is also interesting that chronic supplementation of the long-acting insulin glargin produced improvement in the firstand second-phase insulin secretion in type 2 diabetic patients with mean disease duration of 4.6 years, whereas acute insulin injections reduced glucose-induced insulin response [48]. Harrison and colleagues [49] evaluated ß-cell function in drug-naive patients with newly diagnosed type 2 diabetes prior to and 42 months after treatment with insulin and metformin or a combination of metformin, glyburide, and pioglitazone. As the authors did not find any significant change in Cpeptide or C-peptide/glucose ratios during the study, they concluded that ß-cell function could be preserved at least for 42 months by either treatment.
Characteristics of recent studies assessing preservation of ß-cell function in type 2 diabetes by short-term insulin treatment
T a b l e
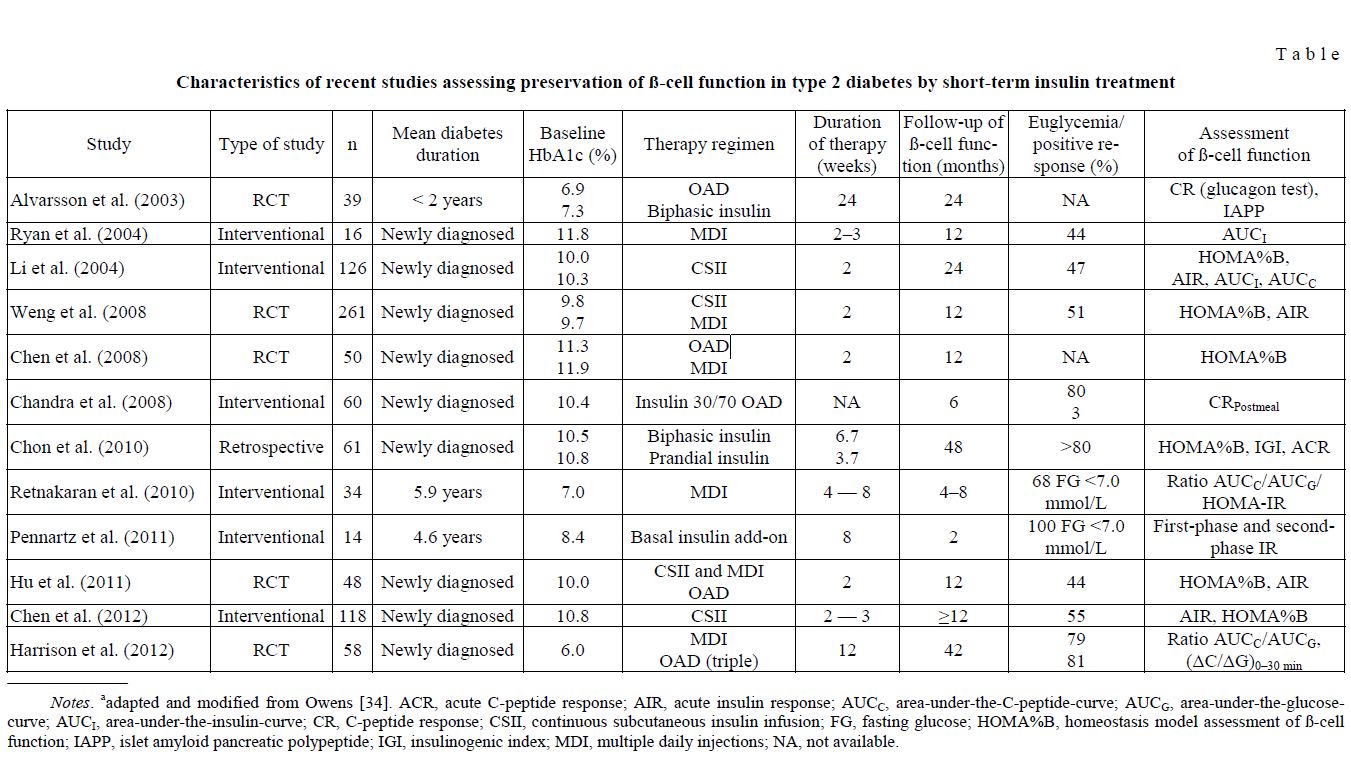
Notes. aadapted and modified from Owens [34]. ACR, acute C-peptide response; AIR, acute insulin response; AUCC, area-under-the-C-peptide-curve; AUCG, area-under-the-glucosecurve; AUCI, area-under-the-insulin-curve; CR, C-peptide response; CSII, continuous subcutaneous insulin infusion; FG, fasting glucose; HOMA%B, homeostasis model assessment of ß-cell function; IAPP, islet amyloid pancreatic polypeptide; IGI, insulinogenic index; MDI, multiple daily injections; NA, not available.
A recent meta-analysis [50] has confirmed that early short-term intensified insulin treatment can improve ß-cell function and decrease insulin resistance. Nonetheless, it must also be recognized that the clinical outcomes among the patients revealed a heterogeneous response. Given the phenotypic heterogeneity of type 2 diabetes, it is conceivable that not all participants in the studies listed in Table, but a variable percentage — on the average 58 % after 1 year [34] — did achieve and maintain a drug-free period of euglycemia, i.e. reversibility of ß-cell function after the short intensified insulin treatment. It is thus most important to figure out the key determinants and mechanisms of improvement in ß-cell function. Kramer and colleagues [51] performed a study in type 2 diabetic patients with mean 3-year disease duration and well-controlled glycemia, using intensive insulin treatment consisting of basal and premeal insulin. They found that baseline HbA1c and change in insulin resistance (HOMA-IR) were independent predictors of reversibility of ß-cell dysfunction. This suggests that elevated glucose concentrations per se (i.e., HbA1c levels) accelerate decline of ß-cell capacity, as displayed in Fig. 1, and that lowering of insulin resistance importantly contributes to the reversibility of insulin secretory function. The pathophysiological basis for early use of insulin has been presented in reviews by Rolla [52] and Joffe et al. [53].
Considering the study outcomes in favor of early intensive insulin therapy, one might ask whether this treatment modality does have negative consequences.
Because of the phenotypic heterogeneity of type 2 diabetes, it is conceivable that not all patients will benefit from early insulin therapy. As pointed out by Lebovitz [54], patients who initially presenting with severe hyperglycemia will take most advantage of it. Intensive glycemic control has been associated with increased incidence of hypoglycemia [55]. However, patients in the Kramer study [51] had very low rates of hypoglycemia (≤ 3.9 mmol/L), which might reflect the moderating contribution of the endogenous insulin reserve, as the authors emphasized. Initiation of insulin therapy in a later stage of disease progression, following failure of oral antidiabetic drugs, requires usually higher doses than in an early stage to control glycemia and thus increases the risk of hypoglycemia and weight gain. Gain in body weight and insulin treatment go often hand in hand and has been shown to be influenced, for example, by baseline HbA1c, the therapy regimen applied, treatment duration, and oral drugs used in combination with insulin. Use of lower insulin doses has the advantage of hampering weight gain [56]. Thus, the dosage used in early short-term intensive insulin treatment is unlikely to provoke weight gain in patients with type 2 diabetes.
Conclusions
Based on pathophysiological evidence, initiation of insulin therapy is evidently the most effective strategy to control glycemia. Insulin has the unique capability of correcting factors involved in the progressive decline of ß-cell function, such as first-phase insulin secretion, insulin resistance, glucolipotoxicity, and inflammation. A number of recent studies have clearly demonstrated that early short-term insulin therapy may modify the disease progression by protecting and restoring ß-cell function. The benefits of insulin therapy are still offered to late, i.e. when ß-cell mass and function are largely lost. It is of utmost importance to preserve ß-cell function in order to maintain good glycemic control to prevent late diabetes complications and improve patients´ quality of life. As the response to short-term intensive insulin treatment is variable, phenotype-targeted therapy may be required to gain the biggest advantage from this intervention. More studies will certainly be needed to verify the current findings and clarify the question whether early intensive treatment with insulin can really alter disease progression by providing long-term protection of ß-cell function.
References
- Butler A.E., Janson J. et al. ß-Cell deficit and increased ß-cell apoptosis in humans with type 2 diabetes // Diabetes. — — Vol. 52, No. 1.— P. 102–110.
- The Diabetes Control and Complications Trial The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus // N. Eng. J. Med. — 1993. — Vol. 329, No. 14. — P. 977–986.
- Turner R.C., Holman R.R. et al. Intensive blood glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33) // — 1998. — Vol. 352, No. 9131. — P. 837–853.
- Del Prato , Bianchi C., Marchetti P. ß-Cell function and anti-diabetic pharmacotherapy // Diabetes Metab. Res. Rev. — 2007. — Vol. 23, No. 7. — P. 518–527.
- Kahn E. The relative contribution of insulin resistance and beta cell dysfunction to the pathophysiology of type 2 diabetes // Diabetologia. — 2003. — Vol. 46, No. 1. — P. 3–19.
- Gastaldelli , Ferrannini E. et al. Beta-cell dysfunction and glucose intolerance: results from the San Antonio metabolism (SAM) study // Diabetologia. — 2004. — Vol. 47, No. 1. — P. 31–39.
- Kohnert D., Freyse E.J., Salzsieder E. Glycemic variability and pancreatic ß-cell dysfunction // Curr. Diabetes Rev. — 2012. — Vol. 8, No. 5. — P. 345–354.
- Robertson R.P., Harmon J., Tran P.O.T., Poitout V. ß-Cell glucose toxicity, lipotoxicity, and chronic oxidative stress in type 2 diabetes // Diabetes. — 2004. — Vol. 53, No. (Suppl. 1). — P. S119–S124.
- Del Guerra , Lupi R., Marselli L. et al. Functional and molecular defects of pancreatic islets in human type 2 diabetes // Diabetes. — 2005. — Vol. 54, No. 3. — P. 727–735.
- Tiedke , Lortz S. et al. Relation between antioxidant enzyme gene expression and antioxidant defense status of insulinproducing cells // Diabetes. — 1997. — Vol. 46, No. 11. — P. 1733–1742.
- Butler A.E., Janson J. et al. ß-Cell deficit and increased ß-cell apoptosis in humans with type 2 diabetes // Diabetes. — Vol. 52, No. 1. — P. 102–110.
- Sladek , Rocheleau G. et al. A genome-wide association study identifies novel risk loci for type 2 diabetes // Nature. — 2007. — Vol. 445, No. 7130. — P. 881–885.
- Steinthorsdottir V., Thorleifsson G. et al. A variant in CDKAL1 influences insulin response and risk of type 2 diabetes // Genet. — 2007. — Vol. 39, No. 6. — P. 770–775.
- Freathy R.M., Timpson N.J. et al. Common variation in the FTO gene alters diabetes-related metabolic traits to the extent expected given its effect on BMI // Diabetes. — 2008. — Vol. 57, No. 5. — P. 1419–1426.
- 15 Spranger J., Kroke A. et al. Inflammatory cytokines and the risk to develop Type 2 diabetes: results of the prospective population-based European Prospective Investigation into Cancer and Nutrition (EPIC) Potsdam Study // Diabetes. — 2003. — Vol. 52, No. 3. — P. 812–817.
- Donath Y., Böni-Schnetzler M. et al. Islet inflammation impairs the pancreatic ß-cell in type 2 diabetes // Physiology. — 2009. — Vol. 24, No. 6. — P. 325–331.
- Yoon H., Ko S.H. et al. Selective ß-cell loss and α-cell expansion in patients with type 2 diabetes mellitus in Korea // J. Clin. Endocrinol. Metab. — 2003. — Vol. 88, No. 5. — P. 2300–2308.
- Böni-Schnetzler , Thorne J. et al. Increased interleukin (IL)-1ß messenger ribonucleic acid expression in ß-cells of individuals with type 2 diabetes and regulation of IL-1ß in human islets by glucose autostimulation // J. Clin. ndocriol. Metab. — 2008. — Vol. 93, No. 10. — P. 4065–4074.
- Donath Y., Halban P.A. Decreased beta-cell mass in diabetes: significance, mechanisms and therapeutic implications // Diabetologia. — 2004. — Vol. 47, No. 3. — P. 581–589.
- Green S., Rozance P.J., Limesand S.W. Consequences of a compromised intrauterine environment on islet function // J. Endocrinol. — 2010. — Vol. 205, No. 3. — P. 211–224.
- Nicolini U., Hubinont C. et al. Effects of fetal intravenous glucose challenge in normal and growth retarded fetuses // Metab. Res. — 1990. — Vol. 22, No. 8. — P. 426–430.
- Wang , Cui Y. et al. Glucose and lipid metabolism in small-for-gestational-age infants at 72 hours of age // J. Clin. Endocrinol. Metab. — 2007. — Vol. 92, No. 3. — P. 681–684.
- Lim L., Hollingsworth K.G. et al. Reversal of type 2 diabetes: normalization of beta cell function in association with decreased pancreas and liver triglycerol // Diabetologia. — 2011. — Vol. 54, No. 10. — P. 2506–2514.
- Kahn E., Haffner S.M. et al. ADOPT Study Group // N. Engl. J. Med. — 2006. — Vol. 355, No. 23. — P. 2427–2443.
- Kendall M., Cuddihy R.M., Bergenstal R.M. Clinical application of incretin-based therapy: therapeutic potential, patient selection and clinical use // Am. J. Med. — 2009. — Vol. 122, No. 6. — P. S37–S50.
- Bunck C., Diamant M. et al. One-year treatment with exenatide improves ß-cell function, compared with insulin glargin, in metformin-treated type 2 diabetic patients // Diabetes Care. — 2009. — Vol. 32, No. 5. — P. 762–768.
- Gudipaty , Rosenfeld N.K. et al. Effect of exenatide, sitagliptin, or glimepiride on ß-cell secretory capacity in early type 2 diabetes // Diabetes Care. — 2014. — Vol. 37, No. 9. — P. 2451–2458.
- Brown B., Nichols G.A., Perry A. The burden of treatment failure in type 2 diabetes // Diabetes Care. — 2004. — Vol. 27, No. 7. — P. 1535–1540.
- Del Prato , Penno G., Miccoli R. Changing the treatment paradigm for type 2 diabetes // Diabetes Care. — 2009. — Vol. 32, No. Suppl. 2. — P. S217–S222.
- DeFronzo R.A. From the triumvirate to the ominous octet: a new paradigm for the treatment of type 2 diabetes // Diabetes. — 2009. — Vol. 58, No. 7. — P. 773–795.
- Kohnert K.D., Hehmke B. et al. Insulin treatment improves islet function in diabetic Chinese hamsters // Exp. Endocrinol. — 2001. — Vol. 109, No. 4. — P. 196–202.
- Absood , Gandomani B. et al. Insulin Therapy for Pre-Hyperglycemic Beta-Cell Endoplasmic Reticulum Crowding // PLoS ONE. — 2013. — Vol. 8, No. 2. — e54351. doi:10.1371/journal.pone.0054351.
- Kobayashi T., Nakanishi K. et al. Small doses of subcutataneous insulin as a strategy for preventing slowly progressive ß-cell failure in islet cell antibody-positive patients with clinical features of NIDDM // — 1996. — Vol. 45, No. 5. — P. 622– 626.
- Owens R. Clinical evidence for the earlier initiation of insulin therapy in type 2 diabetes // Diabetes Technol. Ther. — 2013. — Vol. 15, No. 9. — P. 776–785.
- Rosetti Glucose toxicity: the implication of hyperglycemia in the patho-physiology of diabetes mellitus // Clin. Invest. Med. — 1995. — Vol. 18, No. 4. — P. 255–260.
- Robertson P., Harmon et al. ß-Cell glucose toxicity, lipotoxicity, and chronic oxidative stress in type 2 diabetes // Diabetes. — 2004. — Vol. 53, No. Suppl. 1. — P. S119–S124.
- Monnier L., Colette C. et al. Regulation of oxidative stress by glycaemic control. Evidence for an independent inhibitory effect of insulin therapy // Diabetologia. — 2010. — Vol. 53, No. 3. — P. 562–571.
- Ryan E.A., Imes S., Wallace C. Short-term intensive insulin therapy in newly diagnosed type 2 diabetes // Diabetes Care. — 2004. — Vol. 27, No. 5. — P. 1028–1032.
- Li Y., Xu W. et al. Induction of long-term glycemic control in newly diagnosed type 2 diabetic patients is associated with improvement of beta-cell function // Diabetes Care. — 2004. — Vol. 27, No. 11. — P. 2597–2602.
- Weng J., Li Y. et al. Effect of intensive insulin therapy on ß-cell function and glycaemic control in patients with newly diagnosed type 2 diabetes: a multicentre randomized parallel-group trial // — 2008. — Vol. 371, No. 9626. — P. 1753–1760.
- Chon , Oh S. et al. The effect of early insulin therapy on pancreatic ß-cell function and long-term glycemic control in newly diagnosed type 2 diabetic patients // Korean J. Intern. Med. — 2010. — Vol. 25, No. 3. — P. 273–281.
- Retnakaran R., Yakubovich N. et al. The response to short-term intensive insulin therapy in type 2 diabetes // Diabetes Obes Metab. — 2010. — Vol. 12, No 1. — P. 65–71.
- Chen , Huang Z. et al. Attitudes toward diabetes affect maintenance of drug-free remission in patients with newly diagnosed type 2 diabetes after short-term continuous subcutaneous insulin infusion treatment // Diabetes Care. — 2012. — Vol. 35, No. 3. — P. 474–481.
- Hu , Li L. et al. Short-term intensive therapy in newly diagnosed type 2 diabetes partially restores both insulin sensitivity and beta-cell function in subjects with long remission // Diabetes Care. — 2011. — Vol. 34, No. 8. — P. 1848–1853.
- Chandra T., Priya G. et al. Comparison of gliclazide with insulin as initial treatment modality in newly diagnosed type 2 diabetes // Diabetes Technol. Ther. — 2008. — Vol. 10, No. 5. — P. 363–368.
- Alvarsson M., Sundquist G. et al. Beneficial effects of insulin versus sulfonylurea on insulin secretion and metabolic control in recently diagnosed type 2 diabetic patients // Diabetes Care. — 2003. — Vol. 26, No. 8. — P. 2231–2237.
- Chen S., Wu T.E. et al. Beneficial effects of insulin on glycemic control and beta-cell function in newly diagnosed type 2 diabetes with severe hyperglycemia after short-term intensive insulin therapy // Diabetes Care. — 2008. — Vol. 31, No. 10. — P. 1927–1932.
- Pennartz , Schenker N. et al. Chronic reduction of fasting glycemia with insulin glargin improves firstand second-phase insulin secretion in patients with type 2 diabetes // Diabetes Care. — 2011. — Vol. 34, No. 9. — P. 2048–2053.
- Harrison B., Adams-Huet B. et al. Beta cell function preservation after 3.5 years of intensive diabetes therapy // Diabetes Care. — 2012. — Vol. 35, No. 7. — P. 1406–1412.
- Kramer C.K., Zinman B., Retnakaran R. Short-term intensive insulin therapy in type 2 diabetes: asystematic review and meta-analysis // Lancet Diabetes Endocrinol. — 2013. — Vol. 1, No. 1. — P. 28–34.
- Kramer K., Choi H et al. Determinants of reversibility of ß-cell dysfunction to short-term intensive insulin therapy in patients with early type 2 diabetes // Am. J. Physiol. Endocrinol. Metab. — 2013. — Vol. 305, No. 11. — P. E1398–E1407.
- Rolla The pathophysiological basis for intensive insulin replacement // Int. J. Obes. — 2004. — Vol. 28, No. Suppl 2. — S3–S7.
- Joffe , Freed S. et al. Early use of insulin to improve ß-cell preservation // http://www.diabetesincontrol.com/articles/64-/11534.
- Lebovitz E. Insulin: potential negative consequences of early routine use in patients with type 2 diabetes // Diabetes Care 2011. — Vol. 34, No. Suppl. 2. — P. S225–S230.
- Bonds D.E., Miller M.E. et al. The association between symptomatic, severe hypoglycaemia and mortality in type 2 diabetes: retrospective epidemiological analysis of the ACCORD study // BMJ. — 2010. — Vol. 340. —
- Rosenstock J., Jelaska A. et al. Improved glucose control with weight loss, lower insulin doses, and no increased hypoglycemia with empagliflozin added to titrated multiple daily injections of insulin in obese inadequately controlled type 2 diabetes // Diabetes Care. — 2014. — Vol. 37, No. 7. — P. 1815–1823.