Бірқатар гендердің мутациясына байланысты зат алмасуыны; бузылуымен жүретін түқымқуалайтын аурулар сирек кездеседі, бірақ олар ауыр мүгедектікке, өлімге әкелетін зардаптар қатарына жатады. Солардың бірі мукополисахаридоздар (Mucopolisacharidus + -Qsis+), гликозаминогликан зат алмасуына (қышқыл мукополисахаридтер) катысатын лизосомалды ферменттердің генетикалык кемдігінен болатын, дәнекер тіндердің түқым куалайтын топтағы ауруларына кіреді. Аутосомды-рецессивті типпен түқым қуалайды. Бүл топтағы ауруларды бірінші рет 1917 жылы Гурлер жазған. Дегенмен, бүл аурудың этиологиясы, патогенезі толық шешілмеген. Көрсетілген ферметтердің кемістіктеріне байланысты гликозаминогликан ағзалар мен тіндерде жасушаішілік лизосомдардың бүзылауынан пайда болған өнімдер көптеп жиналады, сондықтан мукополисахаридоздар жинақталу ауруларына жатады.
Нәтижесінде әртүрлі ағзалар мен жүйелердің функциялары бүзылады. Гликозаминогликан дәнекер тіндердің де қурамына кіретін болгандыктан, қаңқалардың жүйелі зақымдалуы, физикалык өсудің тежелуі МПС-ң басты жетекші белгілері болып табылады, әсіресе I-S, IV және VI типтердегілерінде. Аурудың көптеген клиникалык көріністермен жүруі, патологиялык процесске барлык ағзалар мен жүйелердің катысуы, зардаптың адамның әртүрлі жасында басталуы, аурудың диагностикасын киындатады. Сонымен катар, ерте диагносика мен дер кезінде басталған дүрыс терапевтикалық шаралар, әсіресе фермент ауыстырғыш терапия негізгі патологиялык процессті тежеп, наукастарды әлеуметтік жағдайға бейімдеп, өмірінің сапасын біршама жаксартып, тұрақтандырып, пациенттердің өмірін үзартуға ықпал жасайды. Қәзіргі кезде жинақталумен жүретін көптеген лизосомалды аурулар белгілі.
Жіктелуі. Сүйек өзгерістерінің айкын көріну дәрежесі мен психикасының бүзылуы, сонымен катар алмасу бүзылысының тез өршуіне карай, мукополисахаридоздардың негізгі жеті түрін ажыратады. J түрі - Гурлер синдромы. Аутосомды - рецессивті жолмен түқым куалайды. II түрі - Гунтер синдромы. Рецессивті түқым куалайды, жыныспен тіркескен. III түрі - Санфилиппо синдромы. Ауыр жарыместік. Аутосомды - рецессивті жолмен түқым куалайды. Несепте гепаритинсульфат мөлшері көп. IV түрі —Моркио синдромы. V түрі —Шейе синдромы. VI түрі —Марото-Лами синдромы. VII түрі - р-глюкуронидазаның жеткіліксіздігімен негізделеді. МПС-ң әртүрлі синдромдарының сипаттамасы.
Таблица 1
|
Синдромы |
Энзимнің дефектісі |
Хромосомдылык дефект түқым куалайтын түрі |
Клиникасы, лабораториялык мэліметтер |
|
Hurler (МПС-1-Н) |
a-L-идуронидаза |
4 хромосом , аутосомды- рецессивті түрі |
Балалар 6-10 жасында жүректік жэне өкпелік декомпенсациясынан кайтыс болады. Несепте гепаран- жэне дерматан-сульфаттардың деңгейі жогарылаган. |
|
Sheie (МПС-1-S) |
a-L-идуронидаза (толыкемес) |
4 хромосом , аутосомды- рецессивті түрі |
Жүмсақ варианты, калыпты интеллект жэне статура. 30 жаска дейін өмір сүреді. Несепте гепаран- жэне дерматан-сульфаттардың деңгейі жогарылаган. |
|
Hunter (МПС-2) |
Сульфоидуронат сульфатаза |
Х-хромосоммен тіркескен |
Ер балалар ауырады. Аздап акыл-есінің дамуы темендеген. 30-40 жаска дейін өмір сүреді. Кездің мөлдір кабыгы бүлыңгырланбаган. Дорзолюбалды бүкірлік жок. Өршімелі сипатты гирсутизм тэн. Несепте гепаран- жэне дерматан-сульфаттардың деңгейі жогарылаган. |
|
Sunfilippo (МПС-3) |
А түрі — сульфамидаза В түрі— b-N-ацетил- люкозаминидаза С түрі — ацетил-КоА- глюкозамин-N- ацетилтрансфераза Д түрі — a-N-ацетил- глюкозамин-6-фосфатаза |
12 жэне 17 хромосомалар, аутосомды- рецессивті түрі |
Интеллектің айкын ретардациясы, макроцефалия, тэртібінің ете агрессивті болуы. Салыстырмалы элсіз қаңқа езгерістері. Мектеп жасында висцеромегалия пайда болады. Клиникалык типтерінің дифференцацияланбауы мүмкін. Несепте — гепаран. |
|
Morquio (МПС-4) |
N-ацетил-галактозамин- 6-сульфатаза (А түрі) жэне b-галактозидаза (В түрі) |
3 жэне 16 хромосомалар, аутосомды- рецессивті |
Интеллектысы калыпты. Қаңка деформациясы ете катты айкын. Кездің мелдір кабыгы бүлыңгыр. Орталык жүйке жүйесінің зақымдалу дэрежесі аз. Бірінші клиникалык керінісі емірінің 2-3-ші жылында басталып, 20-35 жасында кайтыс болады. Несепте кератансульфат. |
|
Maroteaux- Lamy (МПС-6) |
N-ацетил-галактозамин- 4-сульфатаза |
5 хромосом, аутосомды- рецессивті |
Шамалы бой есуінің тежелуі, айкын бүкірлік, интеллект калыпты, беттің закымдалуы элсіз. Несепте дерматансульфат. |
|
Sly (МПС-7) |
b-глюкуронидаза |
7 хромосом, аутосомды- рецессивті |
Менталды дамулар басымырак, айкын бүкірлік, гепатоспленомегалия, беттің элсіз закымдалуы. Несепте дерматан-сульфат. |
Бақылаауда түрған ауруларға койылған диагноз Maroteaux-Lamy (МПС-6) болғандықтан МПС-ң осы түріне кеңірек тоқталуды жөн көрдік. Мукополисахаридоздың VI түрін (Марото — Лами синдромы, Марото — Лами ауруы) ең алғаш 1960 жылы француз дәрігерлері Марото (P. Maroteaux) және Лами (М. E.J. Lamy) сипаттап жазды. Марото — Лами синдромы —аутосомды-рецессивті жолмен түқым қуалайды. 4-ацетилгалактозамин-4- сульфатаза ферментінің жеткіліксіздігінен тіндерде дерматансульфат жиналады. МПС-ң бүл түрі де баска типтері сияқты бір-біріне, әсіресе I түріне үқсас, бірақ психомоторлы дамудың бүзылысы шамалы немесе мүлдем жоқ. Мукополисахаридоздың 6-шы түрінің клиникалык көрінісі кешірек пайда болады және зардаптың өрбу темпі баяу жүреді. Алғашқы клиникалык көріністер 2 жастан жоғары балаларда пайда бола бастайды. Оған - бой өсуінің артта қалуы, беттің дөрекі көріністері, жоғарғы жақ сүйегінің кіші өлшемі, қысқа мойын, бөшке тәрізді кеуде клеткасы және бұғананың қысқа болуы тән. Қол буындарының бүгу контрактурасы байқалады (наукастар колдарын жоғары көтере алмайды), өсе келе аяқ буындарының контрактурасы қосылады, жүрісі бүзылады. Жиі жедел респираторлы вирусты инфекциялармен ауырады. Жиі жарықтар (грыжа), гепатоспленомегалия анықгалады.
Марото — Лами ауруының симптомдардың айқындалу дәрежесі бойынша типті классикалык (А) және жеңіл, немесе жүмсақ (B), симптомдардың шамалы айқындалуымен көрінетін формаларын ажыратады. Диагнозы клиникалык көріністерге, рентгенологиялык зерттеулердің нәтижесіне, гликозаминогликандардың несептегі экскрециясын анықталуына, жасуша культурасында (тері және лейкоциттердегі фибробластар), пренаталды диагностика кезінде амниотикалык сүйықтықта спецификалық ферменттердің активтілігін зерттеуге негізделеді. Прогнозы, барлык формаларында жақсы емес, себебі өсе келе қаңқа өзгерістері өршиді, әртүрлі ағзалар мен жүйелердің қызметі бүзылады. Профилактикасында медико-генетикалык кеңес және антенаталды диагностика жүргізілуімен, фермент активтілігін және амниотика сүйықтығының жасуша культурасында гликозаминогликандардың қүрамын анықтау көзделеді. Зерттеулерге кіреді: генеалогиялық, клиникалык, биохимиялык, рентген-функциональдық, молекулярлы-генетикалық әдістер.
Емі. Халықаралық тәжірибиеде МПС-пен ауыратын наукастарды емдеу үшін келесі ем түрлері кабылданған: симптоматикалық (медикаментозды, хирургиялык, физиотерапия), алмастыру терапиясы, бағаналы жасушалардың трансплантациясы. Яғни бүл кезде наукастарды әртүрлі мамандар қарайды — хирургтер (жарықты алып тастау), ортопедтер (тірек-қимыл аппаратының бүзылысына ортопедиялык коррекция ), педиатрлар (жиі жедел респираторлы вирусты инфекциямен, жүрек-қантамырлық жеткіліксіздікке және т.б.байланысты), оториноларингологтар (естудің бүзылысы, созылмалы отиттер мен сиуситтер), офтальмологтар, нейрохирургтар және невропатологтар (бас ішілік гипертензия). Емінде гормоналды препараттарды (кортикотропин, глюкокортикоидтар, тиреоидин), А витамины, қан плазма препараттарын кую, декстран енгізу сиякты шаралар тек жағдайының уакытша жаксаруына әкеледі. Физиотерапевтикалык әдістерден закымдалған буынға лидазаның электрофорезі, магнитотерапия, парафиндік аппликация, лазерлі пунктура, омыртка мен буындарға емдік дене шыныктыру, жалпы массаж, мурын-жутқыншак пен ауыз куысының созылмалы ошакты инфекцияларының санациясы колданылады. Баска да емдеу әдістері: антиглаукоматозды операциялар, жарыкты тігу, аденотонзиллэктомия, трахеостомия, гидроцефалия кезінде шунттау, карпалды туннельді синдром себебі бойынша операциялар, жүрек клапандарын, уршык буынын протездеу және т.с.с. Клиникадағы жағдайды келтірейік. Біздің бакылауымызда бауырлас 2000 - жылы туылған 11 жастағы Қ.,және 2001 жылы туылған 9 жастағы Т.,екі ер балалар есепте турады. Анасының айтуы бойынша 11 жастағы Қ., (суретте оң жакта): жасы 1жыл 7 айлығында буындардағы кимылдың шектелгенін, бас сүйегі мен, аяқ-қолдарының өзгергенін байқаған.
Педиатрдің үсынысы бойынша Ташкент қаласындағы ТашМи клиникасында каралып, онда іштен туылған «Жүрек ақауы диагнозына» күмәнденіп, кері кайтарылады. Әрі карай Шымкент каласындағы Облыстык балалр ауруханасының кардиоревматология бөлімшесінде емделіп, жағдайы жаксарып, бірінші рет «мукополисахаридоз» деген диагноз койылып, әрі карай орталык клиникаларда тереңірек тексеріледі. 2001жылы Алматы каласында, сосын генетик-дәрігерлердің кеңесімен Москва каласындағы генетикалык лабороториясында зерттеліп, 2003 жылы глюкозаминогликандардың электрофорездік нәтижелеріне негізделген, МПС-ң VI түрі диагнозы койылады. Зерттелу нәтижелері: глюкозаминогликаннің зәрдегі экскрециясы 64мг/моль креатинин, ферменттер: N-galactozidase-0,98 nmollspot (нормада 0,5-3,2 nmol\spot); а- Iduronidase-0,07 nmol/spot (нормада 0,04-0,4 nmol/spot). 2007 жылы Гамбургтегі Эппендорф университетінің клиникасында диагноз расталып, балалар диспансерлік есепке алынды.
Өмір тарихы бойынша: бала 4-ші жүктіліктен және 4-ші тездетілген босанудан, туылған кездегі салмағы - 4300гр, бойы-53 см, кіндігіне оралып туылған. 3-айға дейін ана сүтімен тамактанған. 3жасында желшешекпен, 4 жасында А вирусты гепатитпен ауырған, генетик пен невропатологтың есебінде турады. Аллергоанамнезінде теріс факторлар жок. Жанұялык анамнезінде. Бала туылған кезде анасы мен әкесінің жастары 20-да болған, дендері сау. Алдыңғы жүктіліктерден туылған үлкен әпкелері бар жоғарғы оку орнында, 11-ші және 8-ші сыныптарда окиды, дендері сау. Баланың әкесінің әпкесі 7-жасында жүре алмай, сөйлей алмай, өзіне-өзі күтім жасай алмаған, белгісіз себептерден кайтыс болған, тексерілмеген, диагноз койылмаған.
Обьективті карағанда: Баланың жалпы жағдайы полиорганды жетіспеушілікке және негізгі ауруына байланысты ауыр. Акыл-есі анык. Жан-жағына адекваты. Жасына сай үй жағдайында 5-ші сыныпта окиды, акылды, компьютрлік сауаттылығы жеткілікті. Буын-тірек аппаратындағы активты кимылдар шектелген,
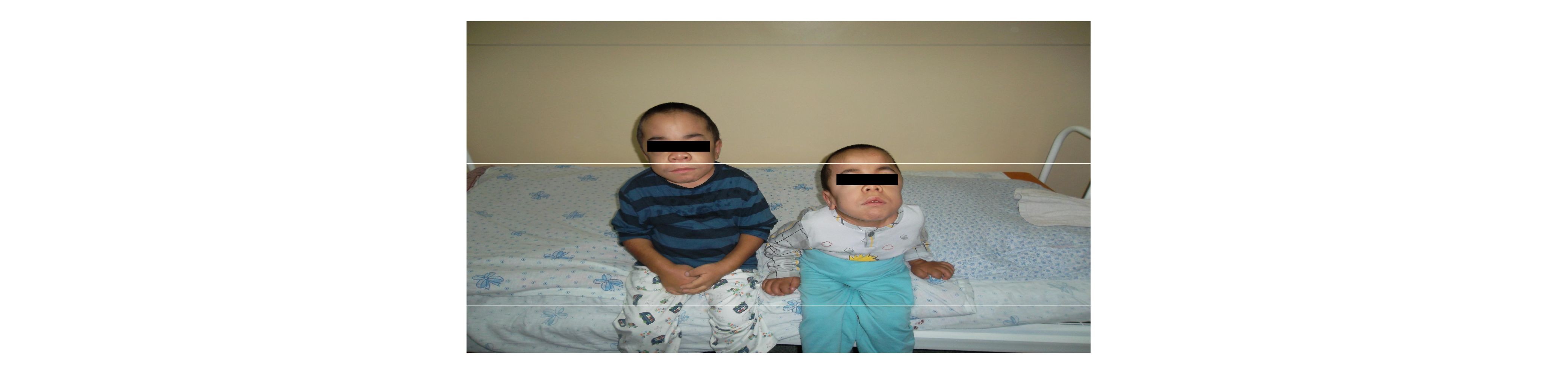
жүрісі «үйректің» жүрісіндей. Баланың физикалық дамуы артта қалған, ергежейлік, бойы -102 см. (қалыптыда - 152 см.), салмағы - 23 кг. (қалыптыда 35-37 кг) және гидроцефалия. Мойны қысқа, маңдай төмпешігі шығыңқы, бас сүйегі деформацияланған, аяқ- қол буындарында контрактуралар, алақандары мен табандары үлкен. Көз алмасы сыртқа шығыңқы, экзофтальмға үқсас, мүрын үсті өте батыңқы.
Көз қарашығы бүлыңғыр, тістері сирек және майда. Көкірек сүйегі «воронка» - қабырғалары «ескек» тәрізді орналасып, кеуде сүйектері деформацияланған, кеуде және бел аймағында кифоздар. Іші үлкен, кіндік айналасының жарығы (грыжа) анықталады. Мүрын арқылы тыныс алуы қиын, тыныстауы шулы, мүрнынан кілегейлі шырыштар бөлінеді. Жүрек тондары түйықталған, жүрек үшында систолалық шу естіледі. Үлкен және кіші дәреттері бүзылмаған Лор дәрігер - Оң жақ есту жолының фурункулезы. Уролог - Зәршығару жолдарының инфекциясы, оң жақ бүйрек толығымен екі есеге үлкейген. Қуықнесепағар рекфлюксының III - дәрежесі. Екіншілік созылмалы пиелонефрит, ремиссия сатысы. Офтальмолог - қарашықтардың дистрофиясы. Қан тобы A (ІІ) Rh оң (+). Жалпы қан, биохимиялық қан, зәр, нәжіс, ИФА, ВИЧ, анализдері қалыпты.
УДЗ - Іш аймағы: аздаған гепатоспленомегалия. Бауыр паренхимасында диффузды өзгерістері. Өт қапшығының гипотониясы. Холестаз. ¥йқы безі паренхимасының реактивті өзгерісі. ЭКГ - Жедел тахикардия, пулсі минутына-120. Қарыншалық, миокард алмасуының бүзылыстары. ЭхоКГ - Жүрекшеаралық және қарыншаралық перделер интактылы, перикардта сүйықтар көрінеді. Қарыншалардың гипертрофиясы. Өкпе гипертензиясының 1-дәрежелі белгілері байқалады. Митралды қақпақшаның алдыңғы және артқы жарғақшалары шашақталған, негізі тығыздалған. РЭГ - Веналық ағысының қиындауына байланысты айқын қан тамырлық гипертензия. Сол жақ қан толымының ассиметриясы 1,0 - 1,2 мм/с (қалыптыда - 1,6 мм/с). ЭЭГ - оң жақ мидың маңдай-алдыңғы самай бөлігінде локалды N- активты биоэнергетикалық белседілігі байқалады.
Қ.-ң клиникалық диагнозы: Мукополисахаридоз VI- түрі (Марото- Лами), екіншілік дилятационды кардиомиопатия, қолқа жетіспеушілігінің 2-дәрежелі регургитация, сол жақ қарынша қуысының кеңеюі. Зәршығару жүйелерінің іштен туылған ақаулары. Оң жақ бүйректің толығымен екі еселенуінің мегауретірдің дамуы, бүйректің созылмалы ауруының 1-дәрежесі. Бүйректің екі жартысы екі еселенген, қуық-несепағар рефлюксының 3-дәрежесі, өршімелі кезеңі. ou- қарашықтың дистрофиясы. Екі жақты диформирленген коксартроз. 9 жасар інісі Т-дағы ерекшеліктер (суретте сол жақта): 1. Ауру тарихы бойынша: Туылғаннан бастап ауру, негізгі ауруға тән клиникалық көріністері 1,5-2 жасында байқалған, дәрігерге басының үлғайғаны және бойының артта қалуымен көрінген. 1 жас, 7 айында диагноз қойылған. 2005 жылы бас миына МРТ жүргізгенде гидроцефалия және бас миының самай бөлігінің екі жақты арахноидалды кистасы анықталған. Сол жағына вентрикулоперинеостомия жасалған. 2. Өмір тарихы бойынша: 5 жүктіліктен, 5 жеделденген босанудан, салмағы - 4300, бойы- 53 см кіндігіне оралып туылған. 3-айына дейін ана сүтімен тамақтанған. Зжасында желшешекпен ауырған, 4 жасында ВГА ауырған, генетик пен невропатологтың есебінде түрады. Бала туылған кезде анасы мен әкесінің жастары 22-де, дендері сау.
Түқым-қуалаушылық сибсы сызығы бойынша: әпкелерінің, бауыры- үлкен ағасының, туысқандарының анамнездері, жағдайлары жоғарыда көрсетілген. 3. Обьективті қарағанда, баланың жалпы, сыртқы көріністері, ішкі ағзаларындағы полиорганды өзгерістердің барлығы ағасына өте үқсас. Т.-ң , бойы - 90 см. (қалыптыда- 118см), салмағы- 16,500 кг.(қалыптыда- 28кг), гидроцефалия. 4. Қан тобы A (ІІ) Rh теріс (-) 5. ЭЭГ - десинхронизирленген жай толқынды - 0 активті, орталық шықпада айқынырақ. Спецификалық эпилептиформалы активтілік, жарты шар аралық ассиметриясы тіркелмеген. Т.-ң клиникалық диагнозы: Мукополисахаридоз vi түрі (Марото-Лами) ОЖЖ тума даму ақауы, окклюзионды гидроцефалия, сол жақ вентрикулоперинеостомиясы. Қарашықтың трофикалық жарасы.Өршімелі, OU қарашық дистрофиясы. Сол жақ сыртқы отит.
Бүл ауруларға Петрии медико-генетикалық орталығында генетикалық зерттеулерге негізделіп МПС vi типті верифицирленген диагноз қойылған. 2004 жылы Москва қаласында және 2007 жылы Гамбург- Эппендорф университетімен дәлелденген. Алматы, Астана және түрғылықты жері бойынша Шымкент қаласының клиникалық стационарында синдромды зерттеуден өткен және симптоматикалық емдер алып түрады. Жүргізіліп жатқан емдеулері: №15-ші емдәм, өмірлік наглазим препаратын в/і, 4 флаконнан инфузомат арқылы аптасына1 рет, цефазалин, актавегин, витамин В6, В1, цераксон, гепадиф, массаж, емдік дене шынықтыру, линекс, Павлов микстурасы, вит - Е, цитраля, пироцетам. Ем реанимация жағдайында жүргізіледі,
175
көрсеткішіне байланысты өкпенің жасанды вентиляциясы (ИВЛ) және шокқа қарсы терапия өткізіледі. Кардиолог, пульманолог, невропатолог, офтальмолог, ортопед, ЛОР. педиатр бацылауларында. Жалпы клиникалық биохимиялық көрсеткіштер (аптасына 1-рет) ЭхоКГ, ЭКГ жүргізіледі.
Сонымен,сирек кездесетін түқым қуалайтын ауру - мукополисахаридозды; VI түрі немесе Маротто- Лами синдромы диагнозын қою спецификалық анамнездер мен фенотиптік көріністер комплексіні; болуына негізделеді, генетикалық зерттеулерді; зертханалық нәтижелерінің (глюкозаминогликанні; зәрдегі экскрециясының көбеюі, N-galactozidase және a- Iduronidase ферменттері белсенділігіні; төмендеуі) дәлелденуі, диагнозды толығымен шешеді. Көрсетілген клиникалық жағдай дәрігер-педиатр үшін де, генетиктер үшін де күнделікті жүмыстарында күтпеген жерден кездесетін сирек ауру. Бүл ауруға диагноз қоюға анамнездері, симптомдардың біртіндеп басталуы, прогрессивті ағымы, бас формасыны; өзгеруі - макроцефалия, бет әлпетінің тек осы зардапқа тәндігі - дисморфия, сүйек-буыннны; деформациялары, гипертрихоз, гепатоспленомегалия, кіндік жарығы, жүректі; қақпақшалық аппаратыны; бүзылысы т.с.с. сияқты белгілер көмектесті.
ӘДЕБИЕТТЕР
- Ахметов М. Медициналыц терминдер сөздігі. -Алматы: Дайк-Пресс, 2009. -800 б.
- Семячкина, А. Н. Мукополисахаридозы у детей / А. Н. Семячкина [и др.] // Рос. вест. перинат. и педиатрии. 2007. № 4. С. 22-29.
- Ashworth, J. L. The ocular features of the mucopolysaccharidoses / J. L. Asworth [et al.] // Eye. 2006. Vol. 20. P. 553-563.
- Clarke, L. A. Idursulfase for the treatment of mucopolysaccharidosis II / L. A. Clarke // Expert Opin Pharmacother. 2008. Vol. 9, № 2. P. 311-7.
- Шабалов Н.П. Детские болезни. Изд. дом «Питер», 2007 г.